
Abstract
Aims: A growing number of genome-wide association studies (GWAS) have revealed associations between single-nucleotide polymorphisms (SNPs) and susceptibility to tuberculosis (TB). However, the results of these studies have been inconclusive. This study evaluated whether the SNPs rs4331426 and rs2057178, identified by GWAS, are associated with TB susceptibility. Methods: We performed meta-analyses for rs4331426, based on eight case-control studies which included a total of 4988 TB cases and 9041 controls; and rs2057178, based on five case-control studies, including a total of 9400 TB cases and 14,459 controls. Results: Our meta-analyses indicated that both rs4331426 and rs2057178 were associated with increased risk of TB (G vs. A: odds ratio [OR] = 1.15, 95% confidence interval [CI]: 1.08-1.22 and A vs. G: OR = 0.84, 95% CI: 0.80-0.88, respectively), especially in an African subgroup. However, no significant TB association was found with rs4331426 in an Asian subgroup. Conclusions: These meta-analyses indicate that rs4331426 and rs2057178 might play a role in the risk of developing TB, especially in Africans; however, rs4331426 might not play a significant role in the risk of developing TB in Asians.
Introduction
T
In the early years of this century, animal experiments suggested that genetic factors, such as toll-like receptor 2 (TLR2) and myeloid differentiation primary response gene 88 (MyD-88), which are involved in innate immunity, might play important roles in the susceptibility to the development of TB (Kramnik et al., 2000; Sugawara et al., 2003a, 2003b). Subsequently, a multitude of candidate gene research studies identified an association between TB susceptibility and genetic polymorphisms in genes such as nucleotide-binding oligomerization domain-containing protein 2 (NOD2), interferon gamma (IFNG), natural resistance-associated macrophage protein 1 (SLC11A1), cluster of differentiation 14 (CD14), and toll-like receptor adaptor protein (TIRAP) (Stockton et al., 2004; Tso et al., 2005; Li et al., 2006; Rosas-Taraco et al., 2007; Nejentsev et al., 2008). Since 2008, several genetic polymorphisms associated with TB susceptibility have been identified by genome-wide association studies (GWAS) (Cooke et al., 2008; Stein et al., 2008; Mahasirimongkol et al., 2009; Thye et al., 2010, 2012). In contrast to methods that specifically test one or a few genetic regions, GWAS scan the entire genome to discover strong associations between single-nucleotide polymorphisms (SNPs) and TB susceptibility.
Among the polymorphisms identified by GWAS, rs4331426 and rs2057178, both located in the noncoding region of the human genome, achieved the highest association scores (rs4331426: odds ratio [OR] = 1.19, p = 6.8 × 10−9; rs2057178: OR = 0.77, p = 2.63 × 10−9). Hence, a number of observational studies have been conducted in different populations to investigate the associations between these SNPs and TB susceptibility. However, the results of different studies have been inconsistent. Therefore, we conducted a systematic review and meta-analysis to get a more precise estimate of the association between these two polymorphisms and TB risk.
Materials and Methods
Search strategy
We searched the PubMed, EMBASE, EBSCO, ISI Web of Knowledge, Chinese National Knowledge Infrastructure (CNKI), and Wanfang (Chinese) databases to identify studies of the association between TB susceptibility and rs4331426 and rs2057178, which were published between January 2000 and July 2015. The search terms were as follows: “tuberculosis” or “TB” in combination with “polymorphism,” “SNP,” “variant,” “mutation,” “allele,” “genotype,” or “haplotype,” and “rs4331426,” “rs2057178,” “genome-wide association studies” or “GWAS.”
Inclusion and exclusion criteria
The inclusion criteria were (1) studies evaluating either rs4331426 or rs2057178 and TB susceptibility; (2) case-control studies of unrelated individuals; (3) studies in which the allele distribution in both cases and controls were available (or data available to calculate them); and (4) studies in which the genotypes of control subjects satisfied the Hardy-Weinberg equilibrium (HWE). Exclusion criteria were (1) studies with insufficient or duplicate data and (2) data from reviews and abstracts. The references of selected articles and reviews were also scanned manually to identify additional eligible studies.
Data extraction
For each retrieved article, data extraction was independently abstracted in duplicate by two independent investigators following the inclusion-exclusion criteria mentioned earlier. The two reviewers solved disagreements by discussion. The major characteristics abstracted from the retrieved studies were the name of the first author, the year of publication, the population of origin, the number of cases and controls, and genotyping methods.
Statistical analyses
The HWE in controls for each study was assessed using asymptotic Pearson's chi-square test before statistical analyses, and p < 0.05 was considered as statistically significant disequilibrium. The association between each polymorphism and TB was estimated by the ORs and 95% confidence intervals (CIs) comparing TB cases to controls for each study. Subgroup analysis was also performed to assess the presence of an ethnic-specific effect. The chi-square based on the Q statistic test and I2 test were used for the assessment of heterogeneity. Fixed-effects models were adopted when p > 0.1; otherwise, random-effects models were used. Sensitivity analysis of the meta-analysis was performed by removing each study step by step to assess the stability of the results. A funnel plot was used to evaluate publication bias. Statistical analyses were carried out using Review Manager 5.1 software (Oxford, England).
Results
Study characteristics
The search of public databases yielded 272 articles; 239 of which were in English and 33 in Chinese. A flow diagram of the selection process is shown in Figure 1. Two hundred sixty articles were excluded by reviewing the titles and abstracts. The remaining 12 articles were reviewed in detail according to the inclusion and exclusion criteria. Supplemental allele distribution data in one article was provided by the authors (Dai et al., 2011). However, seven articles were still eliminated for the following reasons: three articles were reviews, two articles lacked the necessary data, and two articles did not satisfy the HWE in the control group. For articles that included subjects of different ethnic groups or totally independent replication groups, data were extracted for each group (Thye et al., 2010, 2012). One article contained data for both rs4331426 and rs2057178, so this article was separated into two studies (Ji et al., 2013). In total, eight studies for rs4331426 (in four articles) and five studies for rs2057178 (in two articles) were selected for this meta-analysis. Study characteristics are summarized in Table 1. As shown in the table, the included studies were from Ghana, Gambia, Malawi, Indonesia, Russia, and China. Several genotyping methods were used, including the homogenous MassEXTEND assay, the polymerase chain reaction-restriction fragment length polymorphism assay, TaqMan™-based allelic discrimination, or melting temperature (Tm)-shift allele-specific genotyping. The pooled sample size was 14,029 individuals (4988 cases and 9041 controls) for rs4331426 and 23,859 individuals (9400 cases and 14,459 controls) for rs2057178. The allele distributions of the polymorphisms are shown in Table 1.
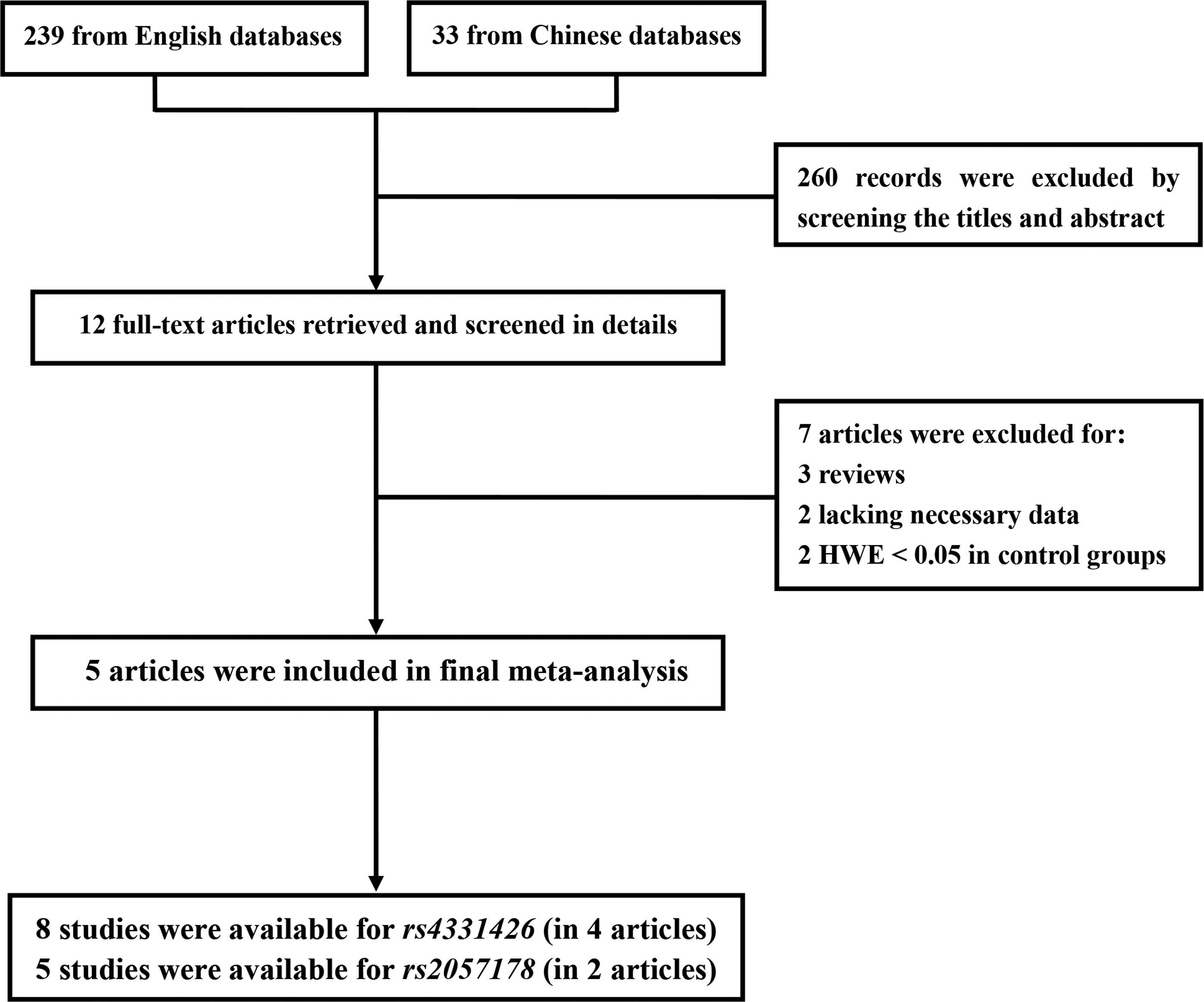
Flow diagram of selection process. HWE, Hardy-Weinberg equilibrium.
95% CI, 95% confidence interval; I, II, independent replication studies I and II in Ghana; Freq, allele frequency; GWAS, genome-wide association studies; MassEXTEND, homogeneous MassEXTEND™ assay; OR, odds ratio; PCR-RFLP, polymerase chain reaction-restriction fragment length polymorphism; TaqMan, TaqMan™-based allelic discrimination; TB, tuberculosis; Tm-shift, melting temperature (Tm)-shift allele-specific genotyping method.
Meta-analysis results
For rs4331426, data were pooled for the allele not the genotype due to the rare frequency of the mutant homozygous genotype of rs4331426 in some included studies. In the final meta-analysis, the G allele was found to be associated with a higher TB susceptibility (G vs. A: OR = 1.15, 95% CI: 1.08-1.22, p < 0.00001; Fig. 2).
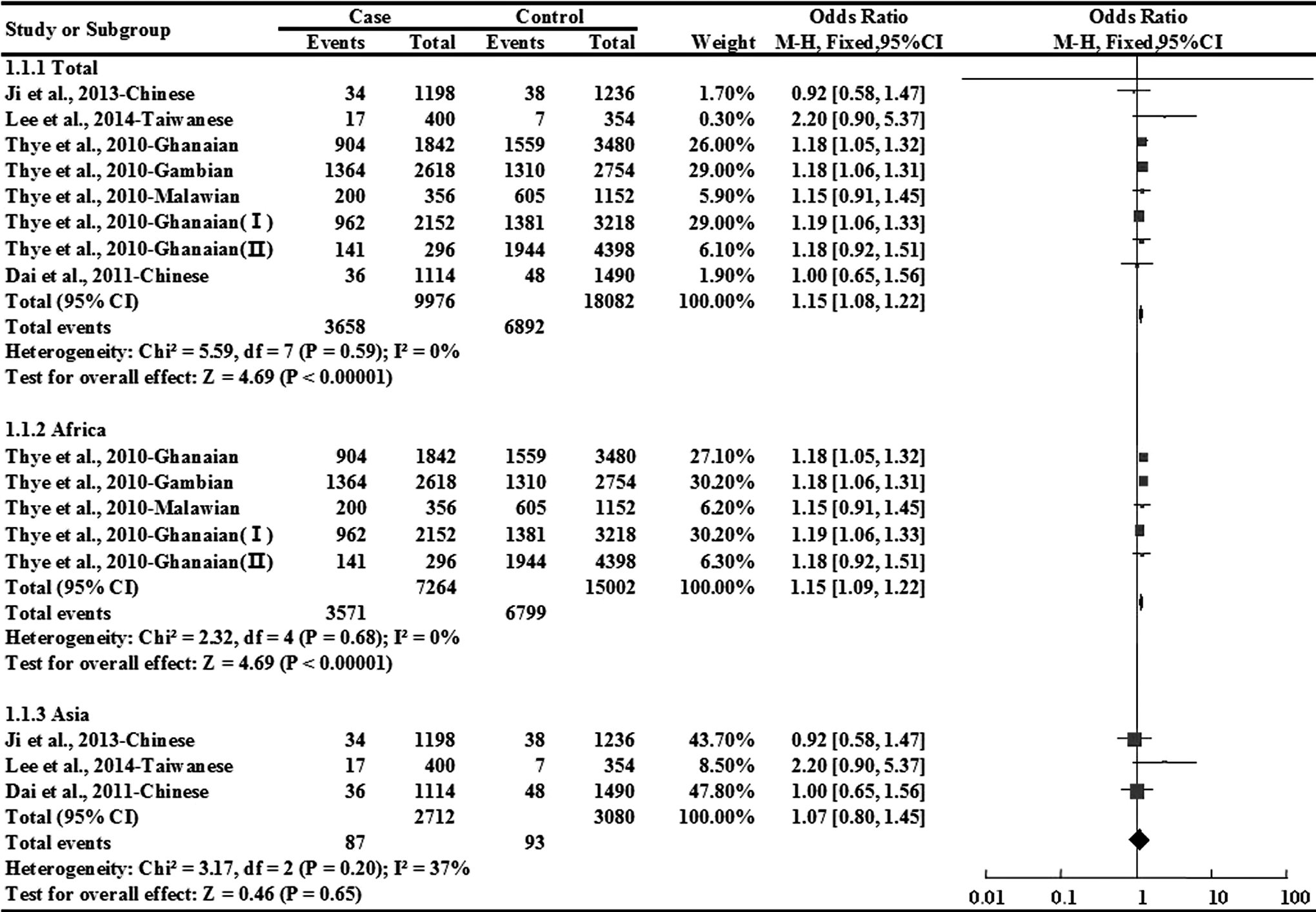
Forest plot of the association between rs4331426 and TB risk in allele comparison. Subgroup analysis was performed by ethnicity. CI, confidence interval; df, degrees of freedom; OR, odds ratio; TB, tuberculosis.
Subgroup analyses were performed to assess ethnic-specific TB susceptibility in Asians and Africans. Significant associations were observed in Africans in the allele model (G vs. A: OR = 1.15, 95% CI: 1.09-1.22, p < 0.00001). However, a significant association was not identified in Asians (G vs. A: OR = 1.07, 95% CI: 0.80-1.45, p = 0.65; Fig. 2).
For rs2057178, the A allele was found to be associated with a lower TB susceptibility (A vs. G: OR = 0.84, 95% CI: 0.80-0.88, p < 0.00001; Fig. 3).
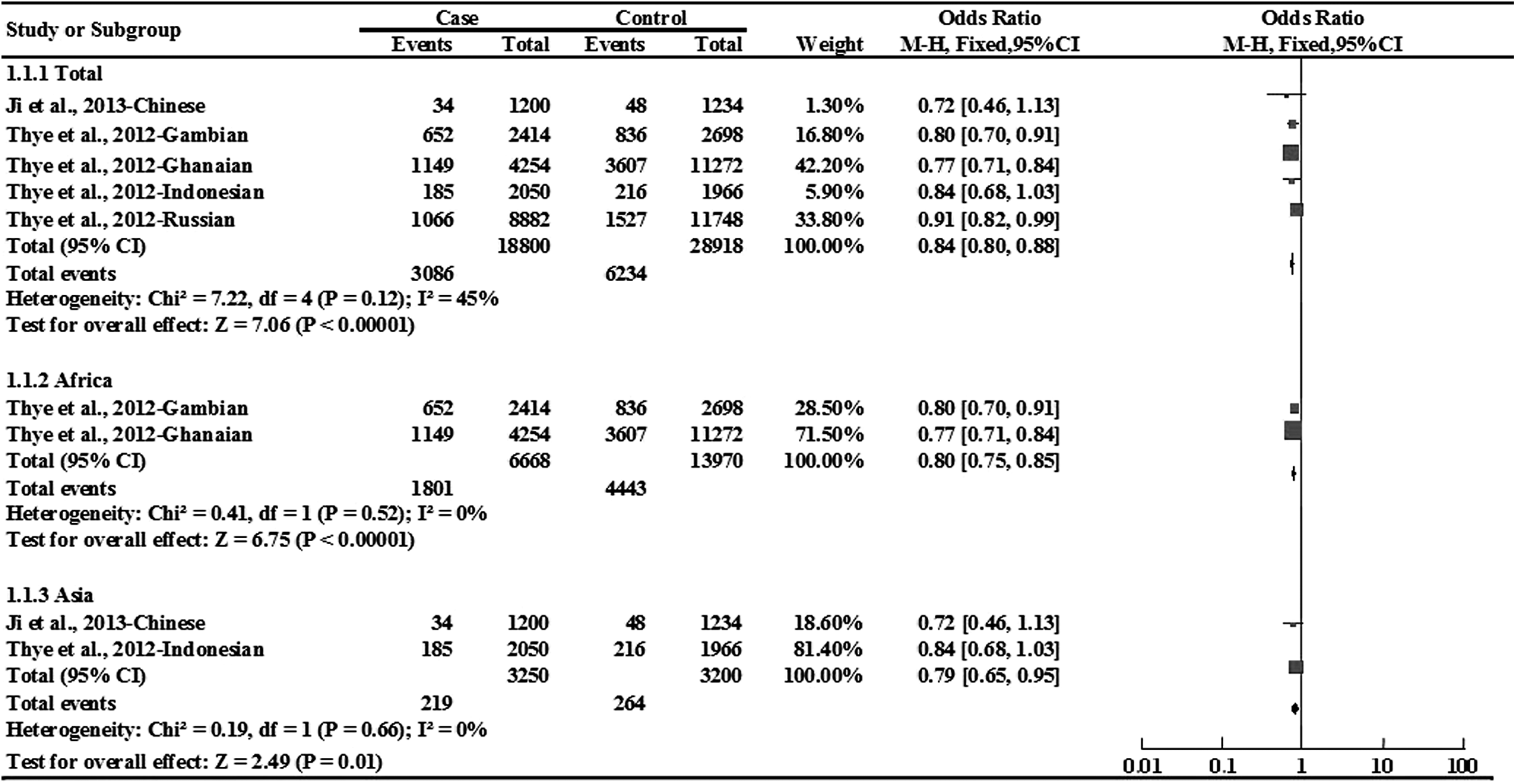
Forest plot of the association between rs2057178 and TB risk in allele comparison. Subgroup analysis was performed by ethnicity.
Subgroup analyses were also performed to assess the ethnic-specific TB susceptibility in Asians and Africans for this SNP. Significant associations were observed both in Africans and Asians (A vs. G: OR = 0.80, 95% CI: 0.75-0.85, p < 0.00001 and A vs. G: OR = 0.79, 95% CI: 0.65-0.95, p = 0.01, respectively; Fig. 3).
Heterogeneity and sensitivity analyses
There was no significant heterogeneity between studies for rs4331426 (pH = 0.59 > 0.1, fixed-effects model), and I2, another index of heterogeneity, was 0%, suggesting no obvious heterogeneity (Fig. 2). There was also no significant heterogeneity between studies for rs2057178 (pH = 0.12 > 0.1, fixed-effects model), and I2 was 45%, suggesting limited heterogeneity (Fig. 3). Individual studies (eight studies for rs4331426 and five studies for rs2057178) in the meta-analysis were sequentially removed to reflect the influence of each study on the pooled OR. For all allele comparisons, the pooled ORs and 95% CIs were not qualitatively different (data not shown).
Publication bias
The shapes of the funnel plots for these polymorphisms were symmetrical in the studies of rs4331426 (Fig. 4), which suggests that there was no significant publication bias. Similarly, no publication bias was detected for the association of rs2057178 with TB (Fig. 4).
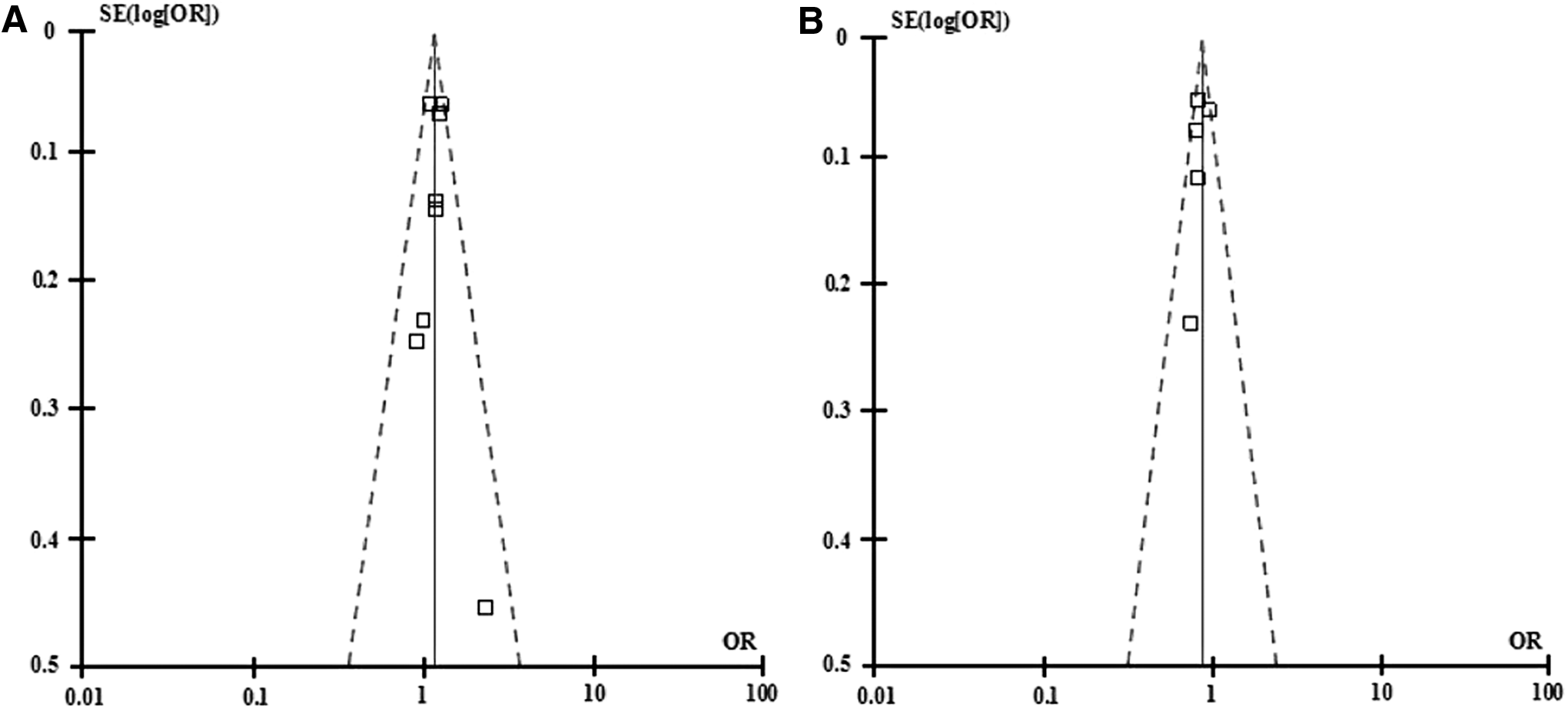
Funnel plot for the relationship between rs4331426 or rs2057178 polymorphism and TB susceptibility in the allele model.
Discussion
Because of conflicting results and low power from relatively few studies, we performed a meta-analysis based on the accumulation of published data to more precisely estimate the association between two SNPs and TB susceptibility. A total of eight case-control studies for rs4331426, including 4988 TB cases and 9041 controls, and five case-control studies for rs2057178, including a total of 9400 TB cases and 14,459 controls, were included in the meta-analysis. Our findings support a role for rs4331426 in the development of TB for the total combined analysis; the G allele carriers had an increased risk of TB compared with those individuals with the A allele. However, sub-group analysis suggested that the rs4331426 polymorphism was a factor increasing risk for TB in Africans, but not in Asians. The present meta-analysis also revealed an association between the rs2057178-A allele and reduced TB risk in both the total combined analysis and subgroup analysis.
The rs4331426 variant is located in a gene-poor region on chromosome 18q11.2. The closest neighboring genes are cutaneous T-cell lymphoma-associated antigen 1 (CTAGE1) and retinoblastoma binding protein 8 (RBBP8), at a distance of 190 and 320 kb to the leading SNP, respectively. rs4331426 was first identified as associated with TB risk in the Ghanaian and Gambian populations (Thye et al., 2010). The other variant, rs2057178, is located on chromosome 11p13, 45 kb downstream of the Wilms' tumor 1 (WT1) gene and at a distance of 500 kb from the reticulocalbin 1 (RCN1) gene. rs2057178 was also first identified as associated with TB risk in the Ghanaian and Gambian populations (Thye et al., 2012). Subsequently, these two SNPs were replicated and tested in additional populations. Our meta-analysis supports the association between rs2057178 and TB risk in Africans and Asians. Our meta-analysis also supports the association between rs4331426 and TB risk in Africans. However, this association might not be present in Asians, such as the Chinese or Indonesian. One possible explanation might be genetic heterogeneity between populations, since the minor allele frequency of rs4331426 was quite different between Asians and Africans (Table 1). Another reason might be different regional adaption and selection processes, such as the environment, pathogen types, and host-pathogen interactions. Therefore, the GWAS of TB risk might be done on Asian and Caucasian populations in further studies.
The GWAS method has successfully pinpointed TB susceptibility-associated variants in the genome in unexpected regions, yielding highly TB-associated hit SNPs in the entire genome based on large population samples. However, one challenge of this method is that associations cannot always be replicated by other research groups or in other populations. GWAS in Thai and Japanese populations were carried out and did not yield any convincing associations, except for finding that several markers might be responsible for TB in young patients (Mahasirimongkol et al., 2012). By using GWAS in the Indonesians and Russians, Png et al. (2012) reported another eight new independent loci that might be associated with TB, within or near immune signaling genes. Unfortunately, these GWAS-identified SNPs also may be inconstant with other studies or have not yet been sufficiently tested in different groups.
For most of the populations, the phenotype of interest was TB, while in a study by Ji et al. (2013), the phenotype evaluated was pulmonary TB. Because the most common TB type was pulmonary TB, which presents to be 90% of all the TB types (Behera et al., 2010; Lawn and Zumla, 2011), we included this article in the meta-analysis. However, if more related articles are published in the future, we will update the meta-analysis by dividing the pulmonary and extrapulmonary TB separately.
The precise biological functions of rs4331426 and rs2057178 are still a puzzle. Like many other disease-associated SNPs identified by GWAS, rs4331426 and rs2057178 are located in “gene desert” regions and the nearest genes are over 45 kb away. These genes are apparently not compatible with functions in pathways relevant to TB. However, one hypothesis is that these are not the causal disease-associated SNPs. Linked SNPs or genes in the neighborhood, as well as a number of, as yet unannotated, open reading frames, may be causal gene loci that are responsible for the TB risk (Meyer and Thye, 2014). Therefore, the next, and possibly difficult, step is interpretation of the biological meaning hidden in these GWAS-identified SNPs.
Footnotes
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NSFC, No. 31001051 and 31302106) and the Natural Science Foundation of Hubei Province (No. 2014CFB230).
Author Disclosure Statement
No competing financial interests exist.