
Abstract
Background: At the cystic fibrosis transmembrane conductance regulator (CFTR) gene (IVS8)-(TG)m(T)n locus, a lower number of thymidines (legacy names 9T vs. 7T vs. 5T) and a higher number of (TG) repeats (TG-11 vs. 12 vs. 13) are associated with decreasing translation of functional CFTR protein in vitro. Methods: Retrospective cohort study comparing phenotypes of California CF newborn screen-positive children (followed 2-8 years) who had two CF-causing mutations (diagnosed as CF) with those who had one mutation from a panel of 40 CF-causing mutations (CF40mut) and one (IVS8)-(TG)11, 12, or 13-5T mutation detected by sequencing (diagnosed as CFTR-related metabolic syndrome [cRMS]). Results: The study included 428 children, of which 234 had two CF-causing mutations, and were used to compare with the other 194 children with one CF-causing mutation and one isolated 5T allele [CF40mut/(TG)13-5T = 21, CF40mut/(TG)12-5T = 85, and CF40mut/(TG)11-5T = 88]. Among children with CF40mut/(TG)13-5T, 38% were diagnosed with CF by 8 years, based on sweat chloride results and clinical presentation. Six percent of those with CF40mut/(TG)12-5T, and none with CF40mut/(TG)11-5T, reached diagnostic criteria. Conclusions: CFTR (IVS8)-(TG)m-5T allele (TG) tract length determination provides valuable information in predicting the risk of developing a CF phenotype. Of the three types of 5T alleles evaluated, screen-positive children with genotype CF40mut/(TG)13-5T progressed from CRMS to CF at a high rate, while there was little evidence of clinical disease in those with CF40mut/(TG)11-5T. Additional data from longer follow-up intervals are needed to fully understand the natural history of individuals with a CF40mut/(TG)m-5T genotype.
Introduction
C
The (IVS8)-(TG)m-(5T) allele is carried by ∼10% of the general population, and it can be inherited by itself or less commonly as part of a complex CFTR allele such as R117H. When in cis with the CFTR R117H allele, R117H penetrance is increased leading to severe lung disease, but often PS (Kiesewetter et al., 1993; Zielenski et al., 1995). The Clinical and Functional Translation of CFTR (CFTR2) mutation database (www.cftr2.org) lists each (IVS8)-(TG)m-(5T) allele as a “mutation of varying clinical consequence,” which means that one of these alleles in trans with a CF-causing mutation may be associated with a phenotype ranging from normal to severe (Sosnay et al., 2013). Our current clinical experience is insufficient to accurately predict the phenotypic risks associated with these alleles.
This study aims to understand IVS8-5T allele penetrance, as a function of the adjacent TG repeats length, by following a population of newborns identified by the California CF newborn screening (NBS) program, which conducts DNA sequencing on newborn blood spots with a single copy of one of 40 CF-causing mutations. An earlier California CF NBS study reported that sweat chloride (SC) and immunoreactive trypsinogen (IRT) concentrations increased in newborns with F508del/(TG)m-5T as “m” increased from 11 to 13 (Keiles et al., 2012). We previously reported on 57 subjects carrying one CF-causing mutation in trans with one or more non-CF-causing mutations (Salinas et al., 2015). This work expands the study population size and follow-up duration of the earlier studies, and focuses on clinical characteristics and risk of CF diagnosis by age 8. Description of the natural history of children in the IVS8-5T subcohort is the focus of this study.
Methods
We used follow-up data retrospectively to describe the natural history of CF NBS-positive newborns identified in the first 4 years of the program. With the California CF NBS algorithm, hypertrypsinogenemic newborns (top 1.6%) are considered screen positive either when two CFTR mutations are detected by a panel of 40 CF-causing mutations (CF40mut) or 1 by the panel and ≥1 by subsequent DNA sequencing (Kharrazi et al., 2015). If on sequencing CFTR (IVS8)-(TG)m-(T)n, poly T status revealed a 5T, TG tract length was reported (Strom et al., 2003; Kammesheidt et al., 2006; Keiles et al., 2012).
Screen-positive infants (considered to potentially have CF, CFTR-related metabolic syndrome [CRMS], or to be CF carriers) were referred to one of 16 California CF centers for diagnostic confirmation, follow-up, and treatment using CF Foundation guidelines (Borowitz et al., 2009). Laboratory and clinical data were entered into the state's centralized web-based database for the initial assessment visits (first 6-12 months of life) until a diagnosis of CF, CRMS, or CF carrier was established and yearly thereafter for CF and CRMS patients. Infants were categorized as CRMS if they (1) had at least two CFTR mutations identified by screening, in which at least one was not classified as CF causing by CFTR2; (2) were asymptomatic; and (3) had an SC <60 mM beyond 6 months of age (Borowitz et al., 2009). A CF diagnosis was given to infants who had (1) at least one SC ≥60 mM; and/or (2) two CF-causing mutations based on the CFTR2 classification. All infants with initial SC <60 mM had a repeat SC test and parental DNA testing to confirm phasing (routinely since July 2011). Personal identifiers were removed and data were analyzed at Children's Hospital Los Angeles (CHLA). The CHLA Institutional Review Board and the California Health and Human Services Agency Committee for the Protection of Human Subjects approved the study and waived informed consent.
The study population was derived from all CF NBS-positive children (n = 848) identified from 2,172,002 births from July 16th, 2007 to July 31st, 2011. The children were followed for 2-6 years with clinical data collected until August 1st, 2013 (initial follow-up period). Those with (IVS8)-(TG)m-5T were stratified into three TG repeat length groups: (IVS8)-(TG)13-5T, (IVS8)-(TG)12-5T, and (IVS8)-(TG)11-5T. Children with (IVS8)-(TG)m-5T known to be part of complex alleles [such as R117H/(TG)m-5T and F311del/1525-42G>A/(TG)12-5T] were excluded. Children with two CF-causing mutations and a CF diagnosis were included for phenotypic comparison. A supplementary set of analyses were conducted comparing the subgroup of CF NBS-positive F508del homozygous children to those with genotype F508del/(IVS8)-(TG)m-5T stratified by TG repeat length. Lost to follow-up was defined as no medical visit for ≥18 months.
SC, PI/PS status, growth parameters, rate of first acquisition of Pseudomonas aeruginosa in the first year of life, and persistent colonization with P. aeruginosa were compared among the four genotype groups [CF40mut/CF-causing, CF40mut/(TG)13-5T, CF40mut/(TG)12-5T, and CF40mut/(TG)11-5T].
SC test
Infants with initial negative SC (<60 mM) had repeat testing at ∼6, 12, and 24 months and later if indicated and the highest value documented.
PI/PS status
Pancreatic status was based on the most recent fecal elastase (FE) value as PI if FE <200 μg/g (Farrell et al., 2008). In the 31% of children missing FE values, pancreatic status was assigned based on use of pancreatic replacement enzymes, as previously described (Sosnay et al., 2013).
Growth
Growth through 12 months was assessed based on weight for height z-scores (WHZ; CDC growth curves from year 2000) for children with a minimum of three growth measurements using all available measurements from each subject to estimate the best fitting curve for each genotype group.
P. aeruginosa status
To assess P. aeruginosa colonization, we documented any positive culture, age at first acquisition in the first year of life, and persistence of colonization. Cultures were obtained during clinic visits by deep throat swabs (Borowitz et al., 2009). Persistent colonization was defined by modified LEEDS criteria: (1) mucoid type, ever; and (2) at least two positive cultures within 12 months (Lee et al., 2003; Pressler et al., 2011). Unknown was used for those with missing data (fewer than two collected cultures per year and/or fewer than 2 years of follow-up).
Clinical diagnosis
Final clinical diagnosis for all (IVS8)-(TG)m-5T subjects was obtained directly from California CF care centers in November 2015, ∼2 years beyond the initial data collection (additional follow-up period; 4-8 years of follow-up). CF clinicians justified the final diagnosis based on SC, microbiology, symptoms, overall clinical course, and imaging reports.
Statistical analysis
Comparisons among the four genotype groups on demographic and clinical characteristics were made with ANOVA or Kruskal-Wallis rank sum tests for continuous variables and chi-square tests for categorical variables. Longitudinal growth data (WHZ) during the first year of life were analyzed using repeated measures mixed models to fit quadratic curves for each group, with CF further divided into PI and PS. Dunnett's post hoc comparisons of least squares estimated means were made with CF-PS children as the reference group. A similar approach was used to analyze sequential SC data, fitting linear curves for each group, with post hoc Tukey-adjusted contrasts for pairwise comparisons of intercepts and slopes. Time to first positive culture for P. aeruginosa during the first year of life was analyzed with the Kaplan-Meier method and groups compared with the log-rank test. Univariate and multiple logistic regression analyses assessed the significance of initial clinical characteristics on CF diagnosis within 8 years. For this analysis, initial SC and IRT were dichotomized at levels associated with CF diagnosis. (TG)11-5T subjects were excluded as none were diagnosed with CF. Statistical analysis was performed with SAS/STAT® v9.2 software. Statistical tests were two sided with a Type I error of p < 0.05.
Results
Population
Of 32,818 hypertrysinogenemic newborns, 848 were considered to be California CF NBS positive. Within this group, we excluded those identified with (1) mutations in cis, (2) CF40mut and a mutation of varying clinical consequence, unknown disease liability, or non-CF causing, (3) CF40mut/R117H/(TG)m-5T (n = 5 of a total of 27 with CF40mut/R117H, which corresponded to 3% of all CF NBS positives), and (4) CF40mut/(TG)m-5T as part of a complex allele (n = 52: 6%). Four hundred twenty eight individuals were analyzed [CF = 234, CF40mut/(TG)13-5T = 21, CF40mut/(TG)12-5T = 85, and CF40mut/(TG)11-5T = 88] (Supplementary Fig. S1 for flowchart; Supplementary Data are available online at www.liebertpub.com/gtmb). The total prevalence of CF40mut/(TG)m-5T was 194 or 23% of all CF NBS positives, with 2.5% (TG)13-5T, 10% (TG)12-5T, and 10% (TG)11-5T.
Compared to the CF group, CF40mut/(TG)m-5T children were more likely to be female (p = 0.0002), have a higher mean birth weight (p < 0.0001), have a lower mean IRT (p < 0.0001), and have a higher lost to follow-up rate (p < 0.0001). The CF40mut/(TG)13-5T group had more Hispanics than the CF group (p = 0.02; Table 1), while the CF40mut/(TG)11-5T group had fewer (p = 0.0001; Table 1).
Denominators are the total subjects per group unless otherwise specified.
The California newborn screening CFTR40 mutation panel consists of the following: 1288insTA, 1717−1G>A, 1812−1G>A, 2055del9>A, 2105-2117del13insAGAAA, 2307insA, 3120+1G>A, 3272−26A>G, 3791delC, 3849+10kbC>T, 3876delA, 406−1G>A, 621+1G>T, 663delT, 711+1G>T, 935delA, A559T, CFTRdele2,3(21kb), F311del, F508del, I507del, G330X, G542X, G551D, G85E, H199Y, N1303K, P205S, Q98R, R1066C, R1162X, R334W, R553X, R75X, S492F, S549N, W1089X, W1204X (3743G>A), W1204X (3744G>A), W1282X.
Children diagnosed with CF based on deleterious genotype (carrying two CF-causing mutations).
Children identified with one panel mutation and a 5T with (TG)13 repeats identified by sequencing.
Children identified with one panel mutation and a 5T with (TG)12 repeats identified by sequencing.
Children identified with one panel mutation and a 5T with (TG)11 repeats identified by sequencing.
p < 0.0001 compared to CF40mut/CF causing.
p = 0.02 for Hispanic versus all others.
p = 0.0001 for Hispanic versus others.
p < 0.05 CF40mut/(TG)13-5T versus CF40mut/CF-causing; p < 0.001 CF40mut/(TG)12-5T versus CF40mut/(TG)13-5T or CF40mut/(TG)11-5T.
CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; IRT, immunoreactive trypsinogen; IQR, interquartile range; SC, sweat chloride.
Sweat chloride
No infants in the CF40mut/(TG)13, 12, and 11-5T subgroups had an initial SC value of ≥60 mM. Maximum SC values were higher in CF40mut/(TG)13-5T compared with CF40mut/(TG)12-5T and CF40mut/11(TG)-5T children (p = 0.0001, Table 1). Mixed model analysis of SC results among TG 11, 12, and 13 subgroups, using a linear fit, showed that SC was significantly related to both increasing age (p < 0.0001) and TG repeat length (p < 0.0001). CF40mut/(TG)13-5T had the highest starting point (α) and the highest rate (β) of increase in SC (Fig. 1). During the first six study years in CF40mut/(TG)12-5T children, none had a maximum SC ≥60 mM, while 33% had an intermediate range of SC (30-59 mM). Among CF40mut/(TG)13-5T children, 15% had maximum SC ≥60 mM and 45% had an intermediate SC (Table 1).

Mixed model analysis for sweat chloride results in (TG) groups: CF40mut/CF causing (black dots, n = 215), CF40mut/(TG)13-5T (green, n = 20), CF40mut/(TG)12-5T (blue, n = 84), CF40mut/(TG)11-5T (red, n = 83). Sweat chloride results were significantly related to age (p < 0.0001) and TG subgroup (p < 0.0001). CF, cystic fibrosis.
PI/PS status
No CF40mut/(TG)m-5T subjects had meconium ileus at birth or PI detected during their follow-up periods (Table 1).
Growth
Growth analysis in the first 12 months showed a significant difference in WHZ scores between CF-PS and CF-PI children (p = 0.03), but no significant difference between CF-PS and (TG)13-5T: p = 0.89, (TG)12-5T: p = 0.97, and (TG)11-5T: p = 0.87 groups (Fig. 2). Secondary WHZ analyses showed significant birth weight effect (p = 0.004), but no independent effect of gender (p = 0.74) or race/ethnicity (Hispanic vs. all others, p = 0.19). Adjustment for birth weight did not alter the comparisons between CF-PS and the other subgroups [CF-PS vs. CF-PI: p = 0.03; vs. (TG)13-5T: p = 0.85, (TG)12-5T: p = 0.99, and (TG)11-5T: p = 0.73].
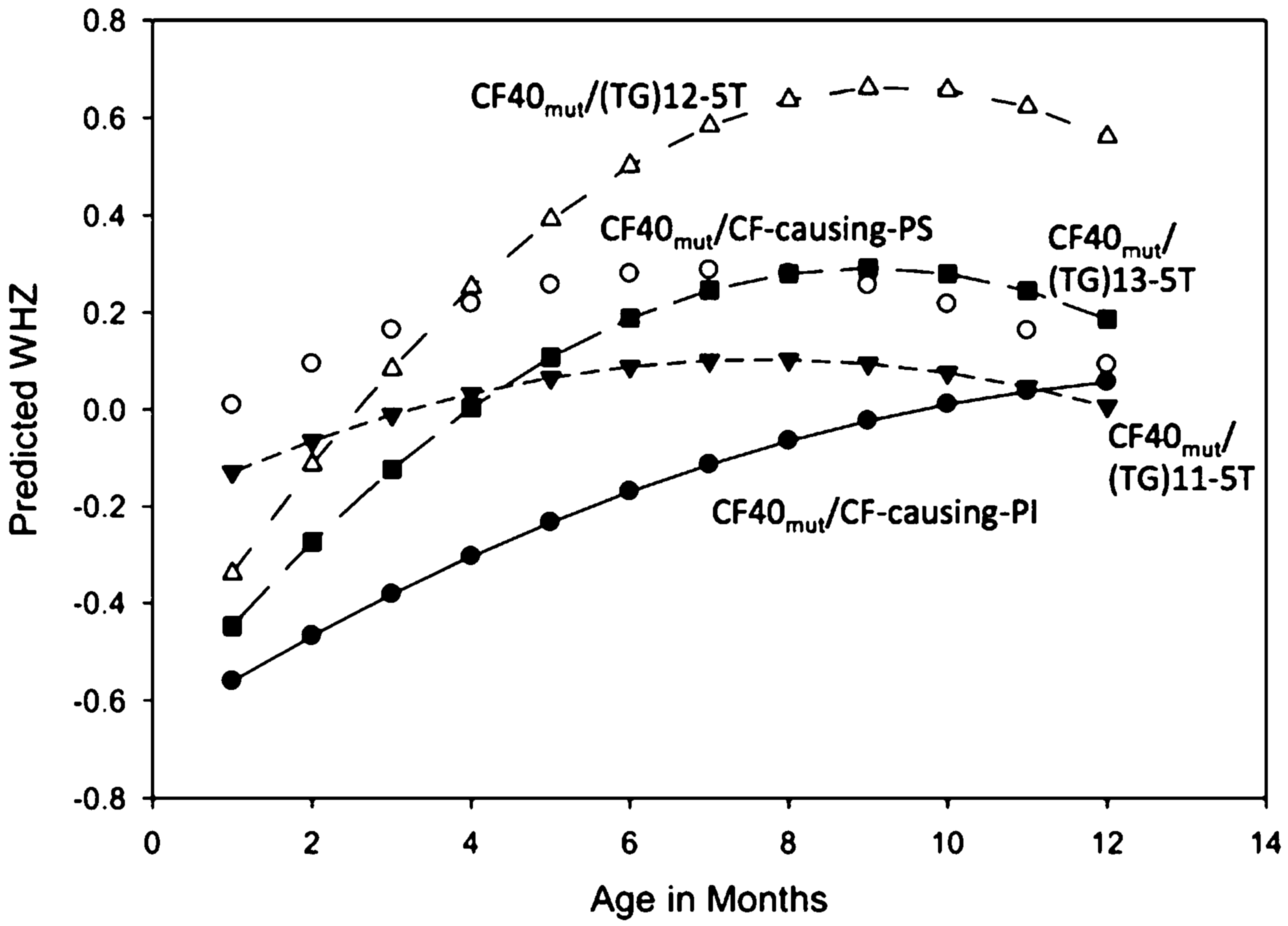
Weight for height Z-scores from initial visit until 12 months in a quadratic model. CF40mut/CF-causing group was subdivided into pancreatic sufficient (PS, open circles, reference) and pancreatic insufficient (PI, black dots); (TG)-5T subgroups were all PS: CF40mut/(TG)13-5T (n = 16, black squares), CF40mut/(TG)12-5T (n = 64, open upward triangles), and CF40mut/11(TG)-5T (n = 36, black downward triangles). Children with <3 growth measurements were excluded. There was a statistically significant difference between CF-PS and CF-PI (p = 0.03), while there was no statistically significant difference between CF-PS and individual 5T subgroups [vs. (TG)13 p = 0.89, vs. (TG)12 p = 0.97, and 11(TG) p = 0.87]. Secondary WHZ analyses showed significant birth weight effect (p = 0.004) and no independent effect of gender (p = 0.74) or race/ethnicity (Hispanic vs. all others, p = 0.15). Adjustment for birth weight did not alter the comparisons between CF-PS and the other subgroups [CF-PS vs. CF-PI: p = 0.03; vs. (TG)13: p = 0.89, (TG)12: p = 0.99, and (TG)11: p = 0.73]. PS, pancreatic sufficient.
P. aeruginosa
First detection of P. aeruginosa in a respiratory culture was assessed through age of 12 months. The probability of acquiring P. aeruginosa was higher among CF40mut/(TG)13-5T compared with CF40mut/CF-causing mutation children (25% vs. 18%, p = 0.0498), CF40mut/(TG)12-5T (0%; p = 0.0006), and CF40mut/(TG)11-5T (4%; p = 0.0033) (Table 1 and Supplementary Fig. S4). There was a higher chronic colonization rate in the CF40mut/TG)13-5T compared with CF40mut/(TG)12-5T and CF40mut/(TG)11-5T groups (no statistical analyses due to missing data; Table 1).
Clinical diagnosis of CF
2 years beyond the initial data collection, three additional CF40mut/(TG)13-5T and two CF40mut/(TG)12-5T children had increased SC, such that final prevalence of SC ≥60 mM was 29% (18%, 52%) (95% CI) and 2% (0%, 8%), respectively, in 4-8 years of follow-up (additional follow-up data). As of November 2015 (age 4-8 years), 8/21 (38%) (18%, 62%) of CF40mut/(TG)13-5T, 5/85 (6%) (2%, 13%) of CF40mut/(TG)12-5T, and 0/88 (0%) (0%, 4%) of the CF40mut/(TG)11-5T children had been converted from the CRMS category to a CF diagnosis. Mean age at CF diagnosis was similar in both (TG)13-5T and (TG)12-5T subgroups (27 ± 24 months). Of the 13 subjects ultimately diagnosed with CF, 5 (38%) were based on clinical justifications alone (e.g., chronic respiratory symptoms, abnormal chest x-ray; Tables 2 and 3).
Supplemental data collected by the California newborn screening program staff up until November, 2015. Physicians were asked to justify the final diagnosis based on DNA results, clinical presentation (clinical), SC in mM, or other.
H.flu, Haemophilus influenza; MRSA, methicillin-resistant Staphylococcus aureus; MSSA, methicillin-sensitive Staphylococcus aureus; PSA, Pseudomonas aeruginosa.
F508del subanalyses: 61% of CF40mut/(TG)m-5T children carried F508del as the CF40mut. Subanalyses were performed comparing phenotypes of children with F508del/(TG)13, 12, and 11-5T subgroups to F508del homozygous children with results similar to the whole CF40mut/(TG)m-5T cohort (Supplementary Tables S1 and S2 and Supplementary Figs. S2, S3 and S5).
Logistic regression probing the association between initial clinical characteristics and CF diagnosis within 8 years among children with CF40mut/(TG)13 or 12-5T mutations suggests that a CF diagnosis was more likely [OR (95% CI), p] for (1) (TG)13-5T relative to (TG)12-5T [20.7 (5.9-72.3), p < 0.0001]; (2) an IRT >80 ng/mL [6.1 (1.3-28.2), p = 0.02]; (3) an initial SC ≥25 mM [6.3 (1.9-21.4), p = 0.003]; or (4) a Hispanic ethnicity [5.2 (1.5-17.5), p = 0.01]. Gender and P. aeruginosa were not predictors of a CF diagnosis. A stepwise multivariate logistic regression analysis among CF40mut/(TG)13-5T and CF40mut/(TG)12-5T subjects showed that IRT >80 ng/mL and CF40mut/(TG)13-5T genotype were independent predictors of a CF diagnosis [adjusted OR 15.4 (1.7-142.9), p = 0.01 and 10.4 (2.4-43.5) p = 0.001], respectively. The small number of CF diagnoses among CF40mut/(TG)-5T subjects limited further additions to the multivariate model.
Discussion
We clinically followed a large cohort of children with genotype CF40mut/(IVS8)-(TG)m-5T, identified through the California CF NBS IRT/DNA/DNA sequencing three-step screening algorithm. Variations in phenotype and eventual CF diagnosis were associated with different (TG) repeat lengths adjacent to the poly-T tract at the CFTR (IVS8)-(TG)m-5T locus. The 106 subjects with genotypes CF40mut/(TG)13-5T or CF40mut/(TG)12-5T together represented 12.5% of all California CF NBS-positive newborns. After 8 years of follow-up, by an average age of 27 months, 12% were considered to have CF. The percentage of screen positives detected to have genotypes, including a 5T allele, would be lower in populations screened by standard IRT/DNA algorithms (Parad and Comeau, 2005).
Our results support prior in vitro studies of (TG) 11, 12, and 13. Exon 9 splice efficiency, studied in cultured nasal epithelial cells and a CFTR minigene system, revealed exon 9+ transcripts in 82%, 61%, and 54% with (TG)11, 12, and 13, respectively, confirming that exon 9 splicing efficiency decreased as the number of TG repeats increased (Hefferon et al., 2004). A classical CF diagnosis has been described in both late-diagnosed patients with genotype CF-causing mutation/(IVS8)-(TG)13-5T and (TG)12-5T and (IVS8)-(TG)m-5T homozygotes. (TG)11-5T is more commonly seen in individuals with CFTR-related disorders such as congenital bilateral absence of the vas deferens (CBAVD) or pancreatitis, and infrequently in patients with a CF diagnosis (Cuppens et al., 1998; Noone et al., 2000; Hefferon et al., 2004; Farrell et al., 2008). Our findings support those reported by Groman et al. in which 134 individuals with a genotype of CF-causing mutation/5T were evaluated for phenotype and TG repeat length. Ninety-one percent (97/107) of symptomatic individuals (CBAVD or “nonclassic” CF) had a 5T allele associated with (TG)12 and 13-5T, while among asymptomatic individuals, 78% (21/27) had a 5T allele associated with (TG)11-5T. They concluded that determination of TG repeat number provided more accurate prediction of 5T allele pathogenicity (Groman et al., 2004).
An SC concentration ≥60 mM is considered diagnostic for CF (Farrell et al., 2008). The SC test is less sensitive and less specific in reliably identifying CFTR dysfunction in individuals with nonclassical presentations (Groman et al., 2005) (Stewart et al., 1995; Quinton et al., 2012). In our study, 38% of those ultimately diagnosed with CF had initial SC between 20 and 30 mM. CF40mut/(TG)13-5T and CF40mut/(TG)12-5T had the steepest rate of rise of SC in the first 6 years of life, while SC rise in (TG)11-5T children was closer to the ∼1 mM /year increase described in healthy 5-10-year-old children (Mishra et al., 2008).
All CF40mut/(TG)m-5T subgroups and CF-PS children had normal growth, as assessed by WHZ. Pancreatic status, not genotype, was associated with growth patterns. That all CF40mut/(TG)m-5T subjects were PS is consistent with current observations in CF-causing mutation/(IVS8)-(TG)m-5T cohorts, which is that this genotype is characteristically linked to a milder form of CF and prolonged survival (Hodson et al., 2008).
Higher than normal rates of P. aeruginosa colonization (Carlson et al., 2009; Rosenfeld et al., 2012) among CF40mut/(TG)13-5T relative to CF40mut/(TG)12-5T and CF40mut/(TG)11-5T children may be explained by a marginal quantity of functional CFTR that leads to an altered milieu on the respiratory epithelial surface, but it is not clear why this rate is higher than those children with two CF-causing mutations. As previously described in a population with atypical CF presentation, P. aeruginosa infection was more likely among individuals with a higher SC and two CFTR mutations (Groman et al., 2005). While P. aeruginosa may rarely be detected among CFTR mutation carriers and healthy infants, it is generally a hallmark of the presence of CFTR dysfunction (Groman et al., 2005; Carlson et al., 2009; Rosenfeld et al., 2012).
The results of our study are important for clinicians. Over a third of children with a positive CF NBS and genotype CF40mut/(TG)13-5T were diagnosed with CF by age 8. These infants may appear normal at birth and have normal SC on initial testing. Thus, families of these patients need to be counseled carefully that their children have a significant risk of developing CF, and close follow-up is recommended. Although limited to a small number of CF diagnoses, our logistic regression analysis should remind clinicians that infants with genotype CF40mut/(TG)13-5T and IRT >80 ng/mL must be followed closely. Because only 6% of CF40mut/(TG)12-5T infants and no CF40mut/(TG)11-5T infants developed clinical CF by age 8, parents may be counseled that early aggressive follow-up is not required. Because data are inadequate on later outcomes, pediatricians and families might be informed to consider evaluation by a CF specialist, should chronic abdominal or respiratory symptoms develop. Maintaining contact with (TG)12-5T families is important, either by yearly CF center clinic visits or through the primary care physician.
Differing lost to follow-up rates were observed among CF40mut (TG)m-5T subgroups, raising the possibility of selection bias. Because late development of symptoms would presumably prompt families to return to a CF center for care, impact of this bias on our main findings is likely small. Given the benign clinical picture observed on follow-up, a decision was made by the California CF NBS consortium to discharge CF40mut/(TG)11-5T children from CF center follow-up after July 1st, 2011. Since that date, (IVS8)-(TG)11-5T, if not part of a complex allele, was no longer reported as CF NBS positive. Longer follow-up is required to quantify the risk of CBAVD, pancreatitis, bronchitis, and other late-onset CFTR-related disorders in these individuals.
Other study limitations include the following: missing data, the retrospective design, the web-based system designed by the screening program to evaluate performance (not clinical research), and a short follow-up period to fully understand mutation penetrance.
Mutation panels used by many NBS programs were designed for adult carrier screening. ACMG guidelines designed direct detection of 5T only reflexly if the R117H mutation is detected. CFTR (IVS8)-(TG)12 and 13-5T are prevalent mutations that can result in CF outside of the usual period of time that initial SC testing is performed. As these mutations are not part of commonly used IRT/DNA mutation panels, programs will misclassify a portion of CF NBS-positive infants with one mutation and an initial SC <30 as CF carriers. Our data support the addition of reflex CFTR (IVS8)-(TG)12 and 13-5T mutation testing after one CF-causing mutation is identified through CF NBS.
In conclusion, CF was ultimately diagnosed in about one third of CF NBS positives with genotype CF40mut/(TG)13-5T. Screen-positive children with genotype CF40mut/(TG)12-5T also progressed from CRMS to CF, but at a lower rate. There was little evidence of clinical disease in CF40mut/(TG)11-5T children followed for 4-8 years. It is important to determine the length of the (TG) repeat tract in children identified with a CFTR (IVS8)-(TG)m-5T allele as one of two CFTR mutations identified through either CF NBS or diagnostic testing of symptomatic individuals to better understand the risk of developing disease. Long-term follow-up and additional CFTR functional assays (such as nasal potential difference and intestinal current measurements) should be considered for screen positives carrying a CF-causing mutation with a (TG)12 or 13-5T.
Footnotes
Acknowledgments
This study received support from the National Institute of Health: USC-CTSI (NIH/NCRR/NCATS; Grant # KL2TR000131) and the Division of Pediatric Pulmonology at the Children's Hospital Los Angeles, and Brigham and Women's Hospital NIH/NHGRI/NICHD U19 HD077671. Additional contributions: All members of the California Cystic Fibrosis Newborn Screening Consortium comprising providers from 16 CF care centers in the state contributed with patient data. Doctor Juan Yang abstracted data for this study from the state's screening information system and assisted with getting up-to-date clinical diagnostic data from care centers for the additional follow-up period.
Author Contributions
Dr. D.B.S. and C.A. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. M.K. and R.B.P. are cosenior authors. Study concept and design: D.B.S., C.A., R.B.P., and M.K. Data acquisition and analysis: D.B.S., C.A., S.Y., and M.K. Interpretation: D.B.S., T.G.K., C.A., S.Y., R.B.P., and M.K. Drafting of the article: D.B.S., T.G.K., R.B.P., and M.K. Critical revision of the article for important intellectual content: D.B.S., R.B.P., and M.K. Obtained funding: D.B.S., T.G.K., M.K., and R.B.P. Statistical analysis: C.A.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.