
Abstract
Background and Aims:
The genetic spectrum underlying familial hypercholesterolemia (FH) remains unclear, especially in northeastern China. The aim of this study was to delineate the FH genetic spectrum and identify specific characteristics of FH patients in this region.
Materials and Methods:
The family history, personal medical history, and lifestyle habits of two unrelated patients clinically diagnosed with homozygous FH were recorded. DNA samples of the patients and their relatives were subjected to a newly designed next-generation sequencing panel using an Illumina Miseq platform. Detected variants were annotated and functionally predicted with in silico algorithms, and protein structures were modeled.
Results:
The patients' cholesterol levels were effectively reduced to 33.8% and 17.2% of the original level under conventional ezetimibe and statin treatment. Two pathogenic mutations, W483X and the novel mutation W483G, in the low-density lipoprotein receptor (LDLR) gene were identified. Both patients were heterozygous for the respective mutations. Under a high cholesterol/carbohydrate diet, these mutations could trigger a severe FH phenotype, but both patients responded well to regular medical treatments and dietary control. The W483X mutation results in a premature stop codon, leading to incomplete protein formation. Although the W483G mutation results in translation of the complete protein with no apparent structural difference, it still led to a severe FH phenotype similar to W483X.
Conclusions:
Identification of the novel W483G mutation expands the genetic spectrum of FH. Both mutations cause a severe FH phenotype under certain conditions, suggesting that W483 is important for LDLR function, highlighting potential targets for genetic screening or drug development.
Introduction
Familial hypercholesterolemia (FH) is a lipoprotein metabolism disease caused by mutations in the low-density lipoprotein (LDL) receptor (LDLR) gene and/or several other important genes, and can be inherited in an autosomal codominant manner (Watts et al., 2014). Individuals with homozygous FH (HoFH), that is, carrying more than one of these codominant mutations, or with the rare autosomal recessive form of FH develop cutaneous xanthomas around their elbows, knees, buttocks, and sometimes in the tendons at a very early age, even at birth in some cases (Cuchel et al., 2014). HoFH is characterized by remarkably elevated LDL-cholesterol (LDL-C) levels and premature atherosclerotic cardiovascular disease, which is estimated to affect 1 in 160,000-300,000 people (Cuchel et al., 2014). Most HoFH patients show only a 10-25% reduction in LDL-C levels even under the highest doses of statin treatment (Cuchel et al., 2014), whereas heterozygous FH (HeFH) is a less severe form of the disease and these patients tend to show a better response to statin treatment; however, the risk of coronary artery disease (CAD) is still significantly increased (Cuchel et al., 2014). Current European Atherosclerosis Society guidelines recommend that both HoFH and HeFH should be diagnosed and medically treated as early as possible to reduce the risk of CAD (Cuchel et al., 2014).
Shi et al. (2014) estimated that the HeFH prevalence in China was ∼1 in 200-300 individuals. In 2017, there were ∼108 million residents in the three provinces of northeast China, Liaoning, Heilongjiang, and Jilin, which would suggest that there were 320,000-540,000 and 360,000-675,000 people living with HeFH and HoFH, respectively, representing a significant public health problem. However, FH cases are rarely reported in this region of China, and patients who are diagnosed with FH scarcely undergo genetic testing. One possible explanation for this situation is that most potential FH patients do not have their cholesterol levels measured, or are misdiagnosed with other diseases due to the general lack of awareness of FH. In addition, people living in the northeast of China have some distinct characteristics compared with populations of other provinces of the country, such as a more oily and salty diet owing to the cold weather in winter. Thus, further focus on FH patients living in the northeast of China would be valuable to gain a better understanding of the genetic spectrum of the disease and the phenotype-genotype correlation.
In August 2018, the Department of Laboratory Medicine of the First Affiliated Hospital of China Medical University cooperated with the Anzhen Heart Team of Beijing Anzhen Hospital to conduct a free clinic in Shenyang, the provincial capital of Liaoning province, to screen suspected patients with HoFH and their family members from all over the northeast of China. Among the recruited index subjects, two patients who were initially diagnosed with HoFH attracted our attention in particular because of their outstanding response to conventional medical treatment with ezetimibe and statin (Table 1).
General Information of the Index Patients
CAD, coronary artery disease; CHD, coronary heart disease; LDL-C, low-density lipoprotein cholesterol; N/A, not available; TC, total cholesterol.
In this study, we analyzed the genetic background of these two index patients and their available family members in an attempt to expand the spectrum of FH pathogenic variants in China and gain a better understanding of the phenotype-genotype correlation, which can facilitate improved diagnosis to initiate earlier treatment.
Materials and Methods
Subjects and families
The suspected HoFH families were eligible to attend the free clinic if the baseline LDL-C level of the index subjects were >13 mM and cutaneous xanthomas were developed before the visit, or the index subjects were previously diagnosed with HoFH by any licensed clinician in China. General and clinical information related to HoFH was collected for all individuals who attended the clinic, including the plasma LDL-C levels before and after medical treatments.
The two unrelated index patients from two different families, residing in the same city were invited to undergo genetic testing for FH-related mutations using a self-designed next-generation sequencing (NGS) panel, which was conducted on a volunteer basis. Subsequently, the patients and their family members were invited to undergo a mutation validation test using Sanger sequencing technology. All individuals who underwent the genetic testing signed the informed consent form, and this study was reviewed and approved by the ethics committee of the First Affiliated Hospital of China Medical University and conformed to the tenets of the Declaration of Helsinki.
Next-generation sequencing
Whole blood samples were collected from the index patients and their family members, and the serum was separated. The DNA was extracted from the whole blood samples using the QIAamp Blood Midi Kit (Qiagen) following the official spin protocol described in the user manual.
The DNA samples of the two index patients were purified and prepared using the Truseq Custom Amplicon Kit (v1.5, Panel ID: 125342; Illumina). The NGS panel was designed to target the exon regions of 24 FH-related genes (Supplementary Table S1) using DesignStudio software (Illumina) at the Department of Laboratory Medicine of the First Affiliated Hospital of China Medical University. Details on the preparation and design of this panel will be published separately.
After the libraries were prepared according to the official protocol provided by Illumina, the samples were sequenced on the Miseq NGS system (Illumina) using the Miseq Reagent Kit V3 (600 cycle; Illumina). The data generated were then analyzed using the TruSeq Amplicon application in Basespace Squence Hub (Illumina). All of the called variants were analyzed using ANNOVAR (Wang et al., 2010) to obtain annotation information, including allele frequencies, and functional prediction was performed using in silico predictive algorithms such as SIFT (Kumar et al., 2009), Polyphen2 (Adzhubei et al., 2010), MutationAssessor (Reva et al., 2007, 2011), and MutationTaster (Schwarz et al., 2014). The variants were then classified according to the 2010 American College of Medical Genetics and Genomics guidelines (Richards et al., 2015). If a novel variant was predicted to be pathogenic, damaging, not tolerated, or disease-causing by at least three of the four predictors, the variant was classified as probably pathogenic (class 4). If a variant could cause a frameshift, stop codon introduction, splicing interruption, or has been previously reported as pathogenic, it was classified as pathogenic (class 5).
Mutation validation and cascade screening
All class 4 or 5 variants identified in the two index patients were further verified using the ABI3730 Sanger sequencer (ABI). The regions containing the targeted variants were sequenced using polymerase chain reaction (PCR) with the primers listed in Supplementary Table S2.
After verification of the mutations screened from the NGS panel in the index patients, all of the available family members were invited to undergo the same validation test using the same PCR primers and sequencer.
Molecular structure modeling
The amino acid sequences of identified variants were entered into SWISS-MODEL (Waterhouse et al., 2018) to generate protein structure models based on the structure of the extracellular domain of LDLR (PDB ID: 1N7D.1) (Rudenko et al., 2002) as the template.
Results
Clinical features of the families
Owing to the one-child policy in China, the two index patients were both the only children in their families and the two families were not related to each other at all.
For family HO-1, only the index patient (HO-1-1, female, 26 years old) and her mother (HO-1-2, 52 years old) were willing to participate in the study (Table 2). Patient HO-1-1 reported that none of the relatives from her father's side had dyslipidemia or any cardiovascular problems to her/their knowledge; therefore, they refused to participate in this study. On her mother's side, her grandmother had a cerebral infarction at the age of 50 and again at the age of 55, and passed away at the age of 80 due to acute myocardial infarction (AMI). Her maternal grandfather also died of AMI at the age of 76, but neither grandparent had been tested for their cholesterol levels. Her mother had two sisters and one brother: the first sister (58 years old) once had her plasma total cholesterol (TC) level measured, which was ∼9 mM, and had a heart attack at the age of 50; the second sister (55 years old) was believed to have normal cholesterol levels but had carotid plaque; and the younger brother (49 years old) had a cerebral infarction at the age of 40. None of these family members were able to attend the free clinic and could not participate in the study owing to the long travel distance; however, they were all informed of the genetic results of the index patients, so that they would be able to easily validate their own status at local hospitals.
Characteristics of the Families Included in the Study
For family HO-2, data were obtained for the index patient (HO-2-1, male, 38 years old) along with his mother (HO-2-2, 65 years old), father (HO-2-3, 64 years old), and grandfather (HO-2-4, 83 years old); however, HO-2-4 refused to receive the genetic validation test. In addition, they were not able to reach their other relatives and did not have information on the family medical history. All of the available information is provided in Table 2.
Detection of candidate variants by NGS
The designed panel covered 98% of the exon region of the 24 genes (Supplementary Table S1), including LDLR, APOB, PCSK9, LDLRAP1, ABCG5, and ABCG8. The in silico analyses indicated the existence of two pathogenic variants, c. T1447G and c. G1448A. The results of the in silico predictions and mutation validation tests are summarized in Table 3, together with the untreated cholesterol levels for both families (also see the pedigree in Fig. 1). The two index patients were found to harbor two single nucleotide variants that are close to each other, leading to a stop-gain (W483X) and missense (W483G) mutation, respectively, at the same amino acid position in LDLR. Furthermore, the W483G variant was identified as a novel pathogenic variant that has not been reported previously.
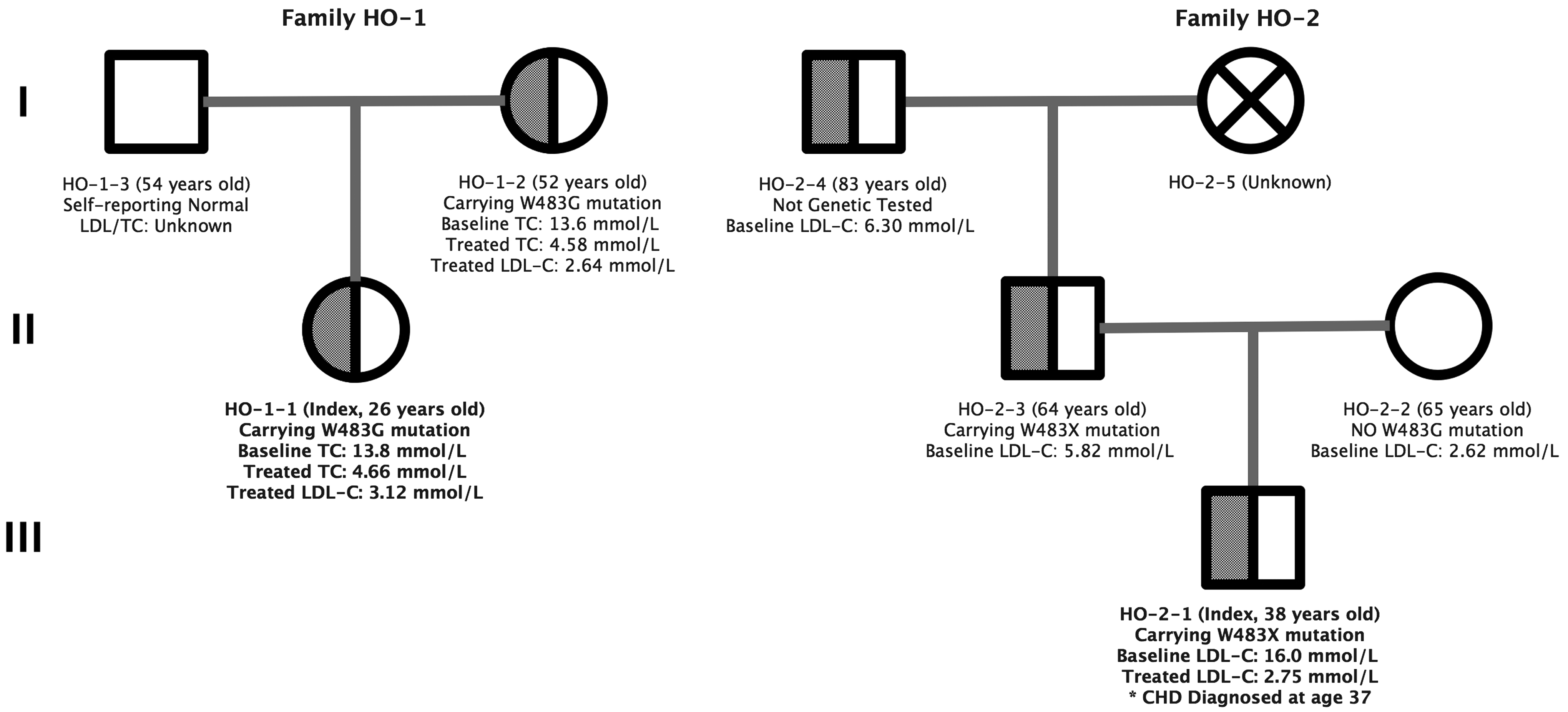
Pedigrees for families HO-1 and HO-2.
Detected Pathogenic Variants
LDLR, low-density lipoprotein receptor.
Patient HO-1-1 and her mother (HO-1-2), both carrying c. T1447G, showed very similar TC levels before receiving any medical treatment (13.8 and 13.6 mM, respectively). Both of their TC levels were reduced to nearly one-third of the baseline level under the regular dose of ezetimibe and statin (Tables 1 and 2), and were still similar (4.66 and 4.58 mM, respectively).
Patient HO-2-1 and his father (HO-2-3), both carrying c. G1448A in LDLR, showed different baseline LDL-C levels (16 and 5.82 mM, respectively), although his mother (HO-2-2) showed a normal LDL-C level (2.62 mM). No potential recessive pathogenic variants were identified in any of the other genes in the panel, such as APOB, PCSK9, or LDLRAP1. In addition, his grandfather (HO-2-4) showed a similar LDL-C level as his father (6.30 mM). Although the grandfather (HO-2-4) refused the mutation validation test, he is highly likely to also be a carrier of this mutation. The LDL-C level of the index patient was reduced to nearly one-fifth of the baseline level under the regular dose of ezetimibe and statins along with aspirin.
Molecular structure model
The structure models were simulated using SWISS-MODEL. As shown in Figure 2, the c. G1448A model clearly demonstrated loss of a portion of the protein structure compared with the wild-type model, with disruption of the conformation immediately before where the tryptophan should appear. However, the c. T1447G model did not show an apparent structural change with respect to either the length of the protein or the tertiary structure.
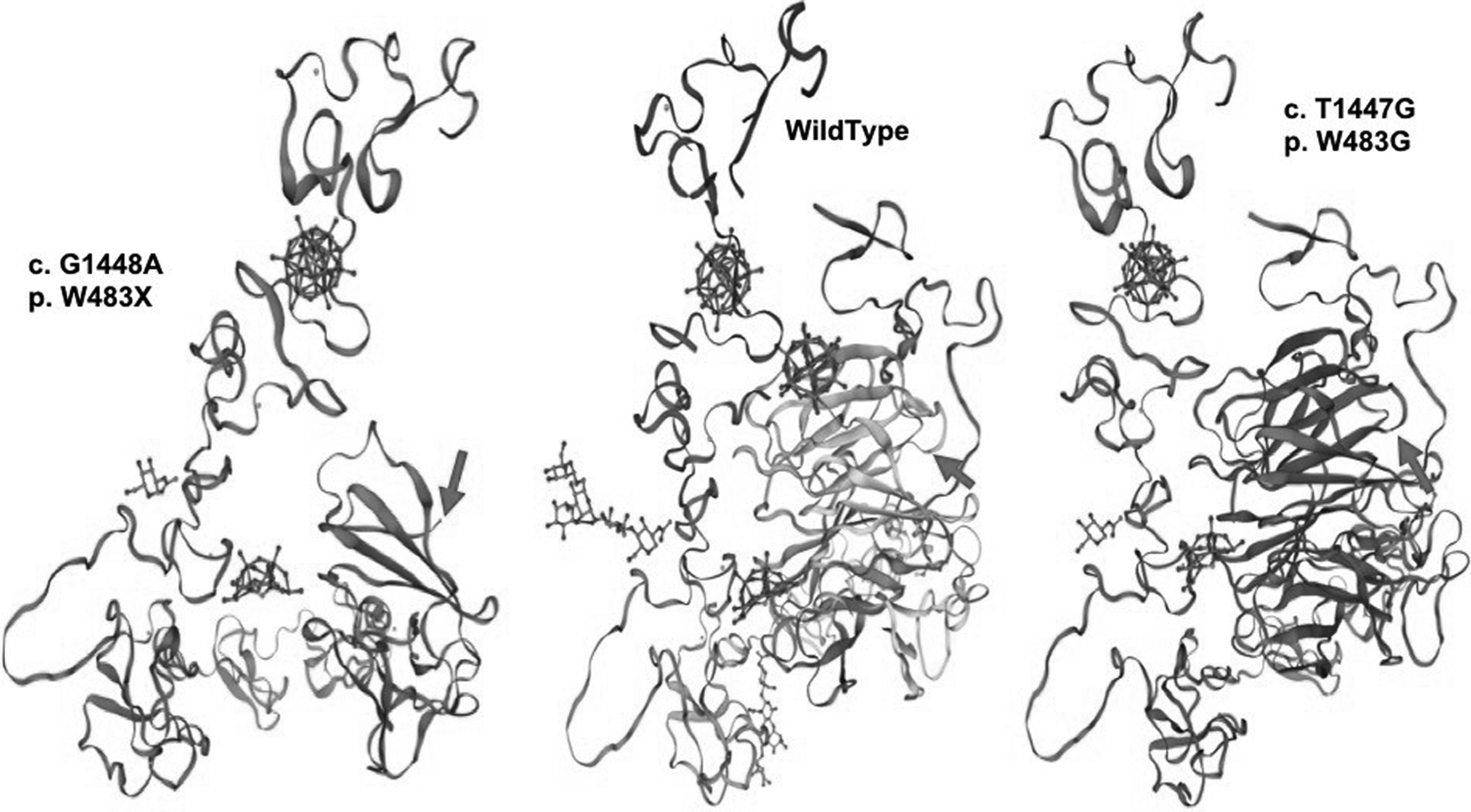
Simulated 3D molecular structure models of the identified variant low-density lipoprotein receptor. The arrows indicate the position of the mutant amino acid.
Discussion
As described in the integrated guidance on FH care published by the international FH Foundation in 2014 (Watts et al., 2014), genetic testing is the “gold standard” method of diagnosis, if a pathogenic variant is detected. In a recent whole-exome sequencing study, including >50,000 individuals (Abul-Husn et al., 2016), only 24% of the FH patients diagnosed by genetic testing could be diagnosed with probable or definite FH according to the Dutch Lipid Clinic Network Diagnostic Criteria (Defesche et al., 2004), the clinical FH diagnostic criteria applied most frequently by clinicians globally. Therefore, the JACC Scientific Expert Consensus Panel, convened by the FH Foundation, concluded that genetic testing could improve FH diagnosis and recommended that FH genetic testing should become the standard of FH care, including the three key FH genes (Sturm et al., 2018). Thus, NGS-based targeted exome sequencing is an efficient tool for FH diagnosis. According to Jiang et al. (2016b), targeted exome sequencing technology could achieve a higher FH pathogenic variance detection rate while saving costs and time compared with whole-exome sequencing. In this study, we applied a self-designed NGS-based targeted exome sequencing method to support the confirmatory diagnosis of FH.
Two patients of northeastern China who were originally clinically diagnosed with HoFH were ultimately found to have a severe form of HeFH after genetic testing and recording of the family history in this study. Specifically, genetic testing revealed two pathogenic single nucleotide variants, c. T1447G (novel) and c. G1448A, which caused different changes at the same amino acid (p. W483) in LDLR. Both variants resulted in a severe phenotype that is similar to that typically observed in cases of HoFH in the two index patients; however, both patients responded well to regular ezetimibe and statin treatment.
The W483X (c. T1448A) mutation was first identified in a patient with FH living in Jiangsu province in southern China (Sun et al., 1994), and was subsequently detected in Austria (Schmidt and Kostner, 2000), Canada (Pimstone et al., 1998), and Italy (Bertolini et al., 2013), although the Canadian case involved a Chinese Canadian family. This mutation has been described as the most common pathogenic mutation in the southern Chinese FH population (Jiang et al., 2015, 2016a) as well as in the general Chinese population (Sun et al., 2018). To our knowledge, this is the first report of the W483X mutation in the northeast of China, indicating that this mutation might have existed earlier and has spread more widely than previously considered.
A functional analysis indicated that the W483X mutation could cause a 17% decrease in LDLR binding activity and a 39% decrease in LDLR internalization activity (Jiang et al., 2016a). Previously reported HeFH patients carrying the W483X mutation showed LDL-C levels ranging from 5.1 to 7.6 mM (Pimstone et al., 1998; Jiang et al., 2016a); however, the index case with this mutation in our study (HO-2-1) showed an untreated LDL-C level of 16 mM, which is much higher than that of all previously reported carriers. Measurement of LDL-C levels of this patient was performed at a general hospital in Shenyang, and his elbow xanthoma appeared during this same period. As described by the patient, right before the first hospital visit, he had been regularly consuming high-cholesterol meals and was continuously drinking excessive amounts of alcohol for months as part of business obligations, which is when his elbow xanthoma appeared. After the hospital visit, he began to attempt to control his cholesterol levels by switching to a low-cholesterol diet solely for ∼1 month before he started to take medications, and his LDL-C level fell to 9.13 mM. Approximately 6 months after he started the medical treatment (10 mg ezetimibe and 10 mg rosuvastatin) along with the low-cholesterol diet, his LDL-C level decreased rapidly to 2.75 mM and his xanthoma also rapidly disappeared.
Although the W483G (c. T1447G) mutation was newly detected in this study, the two patients carrying this mutation (HO-1-1 and HO-1-2) both showed very high untreated LDL-C levels (13.6 and 13.8 mM) compared with those of previously reported HeFH patients. The location of this single nucleotide polymorphism has also been reported for another pathogenic variant (c. T1447C, p. W483R, rs879254905) detected in patients from southern England (Day et al., 1997) and the Netherlands (Fouchier et al., 2005); however, none of these carriers was reported to have such a high LDL-C levels as detected in our index patients. As described by the index patient (HO-1-1), both she and her mother rarely ate meat products but often consumed high-carbohydrate and deep-fried foods such as cakes, cookies, French fries, commercial puffed food, and the like. After starting the medical treatment, they both began a low-cholesterol diet, which led to a rapid decrease in LDL-C levels, and the elbow xanthoma of the index patient also rapidly disappeared. In addition, although her mother (HO-1-2) had a similarly high cholesterol level, she never developed any xanthoma, although the reason for this different manifestation remains unknown.
Based on a review of the literature, we found that position W483, encoded by the 1447th-1449th nucleotides of LDLR, has been reported as a pathogenic variant in multiple forms (Schmidt and Kostner, 2000; Fouchier et al., 2001, 2005; Bertolini et al., 2013; Kong et al., 2015; Sharifi et al., 2016). Pathogenic amino acid substitutions might occur in any of the three coding nucleotides. This suggests that p. W483 could play a critical role in the LDL-C metabolic process. Residue W483 is located immediately behind, but not within, the LDLR repeat class B domain, also known as the YWTD motif, after the most conserved region of the repeat (Marchler-Bauer et al., 2017). The YWTD repeat was predicted to form a beta-propeller structure, as shown in Figure 2. As described earlier, c. G1448A introduced a stop codon to result in a shortened amino acid chain and an incomplete protein molecule. Therefore, it is not surprising that the c. G1448A variant could cause a receptor-negative phenotype as widely reported (Sun et al., 1994, 2018; Schmidt and Kostner, 2000; Cheng et al., 2009; Bertolini et al., 2013), and has been described as the most common FH pathogenic variant in China. By contrast, the novel pathogenic variant, c. T1447G, caused only a substitution from a tryptophan to a glycine residue at this position. Tryptophan and glycine are the two amino acids containing nonpolar and hydrophobic R groups, and thus should be relatively similar with respect to thermal stability. The only notable difference between tryptophan and glycine is that tryptophan is a large amino acid residue, whereas glycine only contains a hydrogen (Fig. 3); however, the mechanism underlying how this difference in the amino acid residue influences LDLR function remains to be elucidated. Such functional investigations could provide a clue or insight for future scientific research or drug development.

Skeletal formula of tryptophan and glycine. The box indicates the difference between the amino acid residues of the wild type and the mutant amino acids.
Taking both cases together and considering their family histories, living habits, and lipid profiles, we suspected that food intake might play a critical role in the abnormal LDL-C levels. Diet counselling has been shown to be beneficial for FH patients (Torvik et al., 2016), and different oil types were also demonstrated to influence the LDL-C levels of FH patients (Negele et al., 2015). However, the specific influence of a high-cholesterol or high-carbohydrate diet on FH has not been investigated to date. Residents of the northeast of China, especially younger residents, are accustomed to eating oily and salty foods, and generally prefer meat over vegetables; moreover, the majority of this population generally lack knowledge about nutrition and regularly consume fast food and delivery food daily. Moreover, due to the lack of FH awareness, most people of this region never have their lipid profiles tested, as this test is not generally included in the public annual health checkup plan. In addition, many cases of high cholesterol, sometimes even for extreme HoFH cases, may not be caused by a single or double inherited condition but rather from a combination of lifestyle choices and the effects of variations in many genes. Given this situation, we suggest that the lipid profile test should be added to the regular health checkup plan, and urge clinicians to pay close attention to young people with unhealthy living habits and high levels of LDL-C, since the cholesterol levels could be more effectively controlled with dietary counselling rather than simply with drugs. Moreover, more food options for a low-cholesterol diet and dietary counselling services should be made more easily accessible for FH patients as needed.
This study has several limitations that should be mentioned. First, we did not perform in vitro or in vivo functional studies for the novel pathogenic variant. Second, we only examined two families with limited family members participating in the study. Third, the assumption about the dietary effect was based on a retrospective analysis from the patients' own descriptions, although the entire process would be impossible to reproduce. The study group will continue to recruit more FH patients and perform more genetic studies to further widen the genetic spectrum of FH in China, especially in the less studied area in the northeast of China. Moreover, we plan to conduct detailed functional studies on the detected variants with cooperation of other laboratories.
In summary, we propose that p. W483 plays an important role in maintaining normal LDLR functions and activities. Further, we have added a novel pathogenic mutation, c. T1447G (p. W483G), to the current genetic spectrum of FH in China. Based on these cases, we believe that a high-cholesterol and/or high-carbohydrate diet can greatly affect the LDL-C levels of FH patients and further influence the risk of cardiovascular disease. Overall, these findings can contribute to improving the current FH genetic screening strategies and help researchers better understand the genotype-phenotype correlation in FH. Further expansion of the genetic spectrum of FH can provide new directions for treatment, management, and drug development.
Footnotes
Acknowledgments
We greatly appreciate the participation of the index patients and their family members in this study. We thank the Anzhen Heart Team for valuable support. Moreover, we extend our gratitude to Dr. Joep Defesche for his help with analysis of the pathogenic variants. The study was supported by two grants (Nos. 81471098 and 81670811) from Natural Science Foundation of China.
Authors' Contributions
S.C. wrote the article, worked on the study concept, and contributed to the data collection, experiments, and data analysis and interpretation. Y.W. and W.W. collected the data and contributed to the analysis and interpretation of data. M.A. and Y.G. conducted the experiments and contributed to the results analysis and interpretation. X.H. contributed to the study concept and article review. L.W. helped with the study concept and reviewed the article. H.S. supervised the whole study and contributed to the study concept and article review.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.