
Abstract
Background:
Familial exudative vitreoretinopathy (FEVR, OMIM 133780), characterized by incomplete retinal vascular development and pathological neovascularization, is a severe inherited retinal disorder. Mutations in 10 genes have been reported to be associated with FEVR, but this still leaves ∼50% of FEVR cases to be genetically explained.
Purpose:
The purpose of this study was to identify novel FEVR-causing mutations and explore the causative mutations in Chinese FEVR families.
Methods:
Whole-exome sequencing was performed to analyze the genomic DNA of the probands from 121 Chinese FEVR families. Sanger sequencing was carried out to verify all identified mutations. Luciferase assays were used to test the activity of a mutant protein in the Norrin-β-catenin signaling pathway.
Results:
Four novel heterozygous TSPAN12 (Tetraspanin 12) mutations, including two single-base substitution mutations and two small-deletion mutations, were identified in these FEVR families: c.1A>G (p.0), c.614G>A (p.G205D), c.695delT (p.V232Gfs*7), and c.833_842del (p.L278Qfs*25).
Conclusion:
This study revealed the causative mutations in four Chinese FEVR families and identified four novel FEVR-causing mutations, thus expanding the mutation spectrum of FEVR in the Chinese population.
Introduction
Familial exudative vitreoretinopathy (FEVR, OMIM 133780) is a severe ophthalmic disease that is mainly characterized by incomplete retinal vascular development and pathological neovascularization. The phenotypes of FEVR vary much among different patients: Vitreoretinal traction, exudates, fibrovascular masses, vitreous hemorrhages, retinal folds, and tractional retinal detachment can be partially or completely found in patients (Canny and Oliver, 1976; Laqua, 1980).
FEVR was first reported in 1969, and it was defined as an inherited retinal disorder (Criswick and Schepens, 1969). In the past half century, several genes and loci have been identified to be associated with FEVR, including NDP (Norrie disease protein), LRP5 (low density lipoprotein receptor-related protein 5), FZD4 (frizzled class receptor 4), TSPAN12 (tetraspanin 12), ZNF408 (zinc finger protein 408), KIF11 (kinesinfamily member 11), CTNNB1 (catenin beta 1), ATOH7 (atonal homolog 7), RCBTB1 (RCC1 and BTB domain containing protein 1), and EVR3 (exudative vitreoretinopathy 3) on chromosome 11p12-13 (Toomes et al., 2005; Junge et al., 2009; Ye et al., 2009; Collin et al., 2013; Hu et al., 2016; Wu et al., 2016; Keser et al., 2017; Panagiotou et al., 2017). Despite this, there are still about 50% of FEVR cases left to be solved (Gilmour, 2015). In addition to various causative genes, the genetic patterns of FEVR patients also show diversity, including autosomal dominant, autosomal recessive, and X-linked pattern (Plager et al., 1992; Chen et al., 1993; Toomes and Downey, 1993; de Crecchio et al., 1998; Jiao et al., 2004; Poulter et al., 2016). Even mutations in the same gene can show different patterns, which posed additional difficulties for molecular diagnosis of FEVR (Jiao et al., 2004; Toomes et al., 2004; Poulter et al., 2012, 2016).
As no effective treatment is available for this disorder at present, coupled with the complexities in clinical and genetic features, molecular diagnosis is of great importance for this disease. In recent years, benefiting from the rapid development of next-generation sequencing (NGS) technology, NGS-based whole-exome sequencing (WES) has become a powerful tool in molecular diagnosis for retinal diseases with multiple pathogenic genes. To reveal the causative mutations in 121 Chinese FEVR families, we carried out WES analysis on the probands of these FEVR families. As a result, four mutations shown in Table 1 (c.1A>G, p.0; c.614G>A, p.G205D; c.695delT, p.V232Gfs*7; c.833_842del, p.L278Qfs*25) in the TSPAN12 gene were identified as probably being responsible for the disease in four families. Further bioinformatics analyses and protein function studies proved the pathogenicity of these mutations. These four mutations are first reported to be associated with FEVR. This study revealed the causative mutations in the four FEVR families, expanded the TSPAN12 mutation spectrum in Chinese FEVR patients, and will provide some guidance for molecular diagnosis of FEVR.
Causative Mutations in the Four Families
Materials and Methods
Subjects and clinical evaluation
This study adhered to the Declaration of Helsinki. All study protocols were approved by institutional ethics committees of Sichuan Provincial People's Hospital and Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine. Written informed consent was obtained from all subjects who participated in this study.
For this study, all the FEVR families were recruited from Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine. All participants were evaluated by a clinical ophthalmologist based on fundus photographic and angiographic changes by fundus fluorescein angiography (FFA). The angiographic examinations were assessed by intravenous injection of fluorescein dye. Premature birth, history of oxygen inhalation, and drug abuse were all excluded.
Mutation screening by WES
Peripheral blood from members of the FEVR families was collected into EDTA anticoagulant tubes and then genomic DNA was extracted by using a blood DNA extract kit according to the manufacturer's protocol (Qiagen, Germantown, MD). DNA was stored at −20°C for use in subsequent analysis. The DNA samples from the 121 probands were subjected to WES as per the following methods. Approximately 1 μg of the genomic DNA sample was sheared into fragments of 200-500 bp in length. Then, the sheared fragments were end repaired; Klenow exonuclease was used to add an A base to the 3ow exon Illumina index adaptors that were ligated to the repaired ends; and finally, eight cycles of polymerase chain reaction (PCR) amplification were applied to each sample. In each capture reaction, 50 precapture DNA libraries were pooled together. Fifty precapture DNA libraries were pooled together in each capture reaction. The targeted DNA was captured by NimbleGenSeqCap EZ Hybridization and Wash kit (NimblegenSeqCap EZ Human Exome Library v.2.0) following the manufacturer's protocols for WES. Captured libraries were then sequenced on Illumina HiSeq 2500 (Illumina, San Diego, CA).
Sequencing data were analyzed by the NextGene V2.3.4 software, and reads were compared with the hg19 human reference sequence from UCSC by BWA. Variants were called by using Atlas2 and noted by ANNOVAR. Variant frequency data were obtained from the dbSNP database, 1000 Genomes, NHLBI Exome Sequencing Project (ESP) (EVS, ESP6500), the ExAC Browser (Beta), gnomAD browser beta, and 2800 in-house non-FEVR controls.
Mutation validation
Sanger sequencing was carried out to confirm the mutations identified by WES. To do this, PCR primers (shown in Table 2) were designed and synthesized by Sangon Biotech (Shanghai, China) to amplify genomic DNA fragments containing the candidate loci. The PCR products were purified by FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific Fermentas), and then were directly sequenced by using BigDye version 3.1 and an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. DNA samples from members of the four families and 500 ethnic-matched control individuals were subjected to Sanger sequencing.
Primers for Sanger Sequencing
Construction of expression plasmids
C-terminal Flag-tagged human TSPAN12 expression cDNA were purchased from Origene, Inc. (Rockville, MD), and they were sub-cloned into pCMV6 expression vectors. Point mutations were introduced into the wild-type pCMV6-TSPAN12 expression plasmid by site-directed mutagenesis using a QuikChange® Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). To construct LRP5 expression plasmid, the cDNA encoding wild-type LRP5 (Origene, Inc.) was sub-cloned in-frame into the pRK5 vector (BD Bioscience, San Jose, CA) by using the XbaI and HindIII sites. FZD4 and NORRIN expression plasmids (generously provided by Dr. Jeremy Nathans of Johns Hopkins University) have been previously described (Xu et al., 2004).
Expression of TSPAN12 plasmids in cells
Western blotting method was used to assess the expression of TSPAN12 plasmids. To do this, 293T cells were cultured in Dulbecco's modified Eagle's medium with high glucose (Gibco) supplemented with 10% fetal bovine serum and 1% (v/v) penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. The cells were seeded into 6-cm plates and then transfected with 3000 ng of pCMV6 vector or 3000 ng TSPAN12 plasmid (wild-type or mutant), respectively, by using the Lipofectamin 3000 transfection reagent. Forty-eight hours later, the cells were lysed in sodium dodecyl sulfate (SDS) lysis buffer (2% SDS and 62.5 mM Tris-HCl, pH 6.8, containing protease inhibitors cocktail tablets ordered from Roche, Inc.) and sonicated three times for 5 s. Equal amounts of protein (20 μg) were loaded to a 12% polyacrylamide gel and analyzed by immunoblotting. The antibodies used for western blot are anti-Flag (Cat# F1804, 1:3000 dilution; Sigma, St Louis, MO) and anti-β-actin (Cat# 20536-1-AP; Proteintech, Chicago, IL). Horseradish peroxidase conjugated goat anti-rabbit secondary antibody (Cat#7074, 1:5000 dilution; Cell Signaling Technology) was used for immunoblotting.
Luciferase assays
To test the protein activity of the mutant TSPAN12 protein, HEK293 cells stably harboring the Wnt/β-catenin reporter SuperTOPFlash (HEK293STF) were seeded into a 24-well plate and then co-transfected with the following expression plasmids: 400 ng of TSPAN12, 200 ng of NORRIN, 200 ng of FZD4, 200 ng of LRP5, and 100 ng of pSV-β-Galactosidase Control Vector (Promega, Madison, WI) for each well by using Lipofectamine™ 3000 Transfection Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The transfected cells were used for luciferase activity assay by using the Promega Luciferase reporter assay system after 48 h. The firefly luciferase activity was normalized to the co-expressed β-galactosidase activity. Each assay was performed in triplicate at the same time and repeated four times.
Results
Clinical evaluation
Clinical information for the four probands and their family members who participated in this study is shown in Table 3. In family 1 (Fig. 1), the proband II:2 was a 3-year-old boy who was diagnosed as FEVR, as well as his mother (I:2) and sister (II:1). The Fundus photography and FFA examination of his mother showed that temporal peripheral ischemia led to neovascularization, dilation of peripheral retinal vessels, and fluorescein leakage in both eyes (Fig. 2A), whereas his father was normal (Fig. 2B). In family 2 (Fig. 1), the proband II:1 suffered ophthalmic problems and presented retinal detachment since birth, whereas her mother had no abnormalities (Fig. 2D). The Fundus photography and FFA examination of her father showed that temporal peripheral ischemia leads to dilation of the brush-like peripheral retinal vessels in both eyes and neovascularization and fluorescein leakage in the right eye (Fig. 2C). In family 3 (Fig. 1), the proband II:1, a 5-year-old girl, was confirmed as having FEVR and presented with mild vitreous opacity. Ophthalmic examination of her mother also showed FEVR conditions, including multi-branch peripheral retinal vessels with a brush-like appearance in both eyes (Fig. 2E). Fundus pictures and FFA examination on her father showed no abnormalities (Fig. 2F). In family 4 (Fig. 1), the proband III:1 developed retinal detachment in both her eyes. The ocular examination of her mother showed that both her eyes exhibited typical straightening of the peripheral vessels with fluorescein leakage (Fig. 2G). Her maternal uncle was also diagnosed as having FEVR, whereas her father was normal (Fig. 2H).
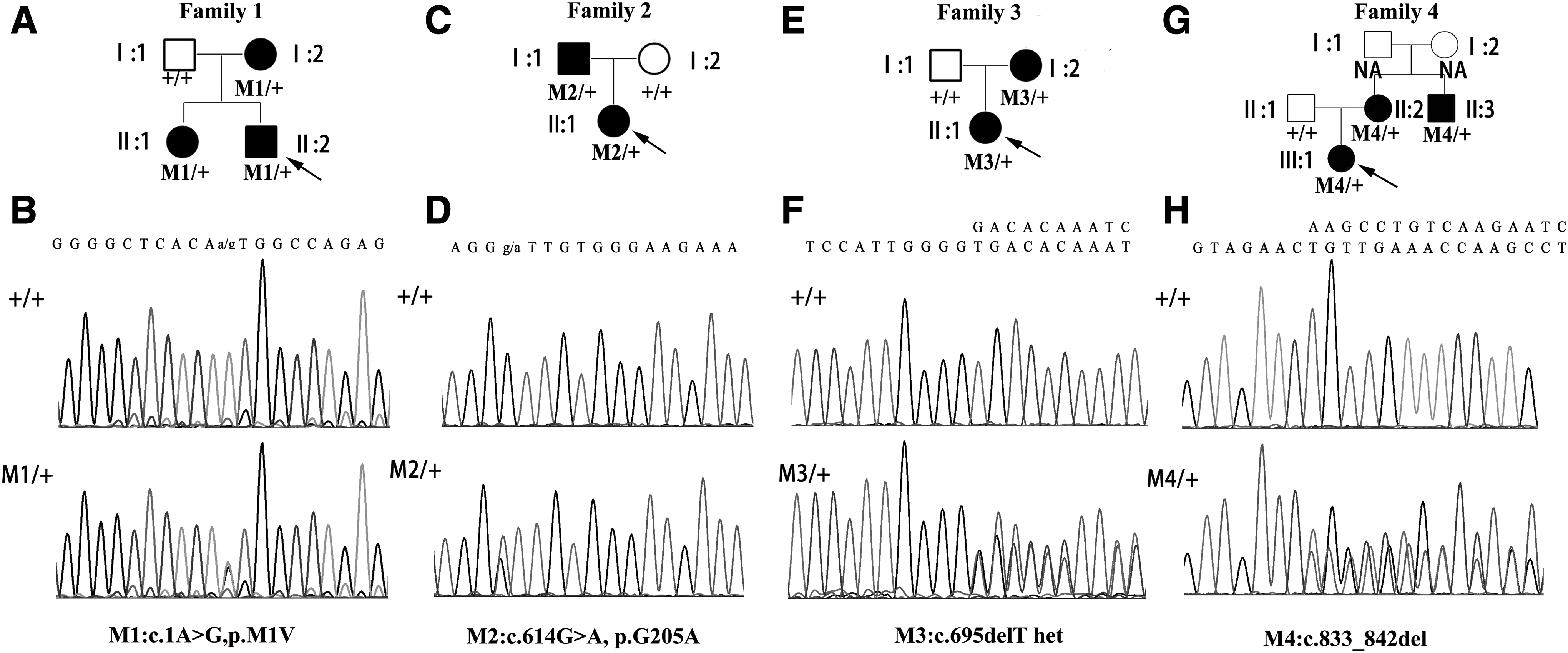
Pedigrees of the four FEVR families with TSPAN12 mutations and corresponding Sanger sequencing chromatograms (
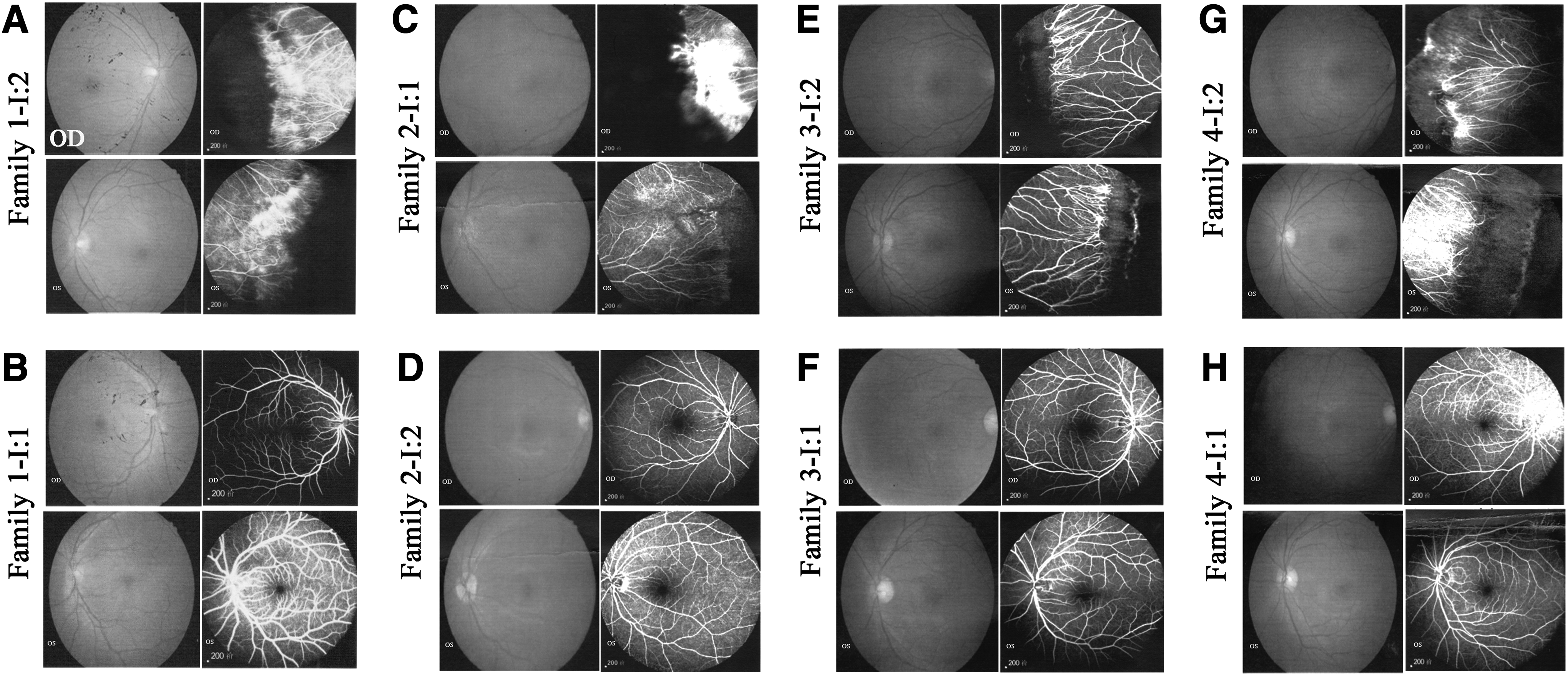
Fundus photograph and FFA of family members from the four FEVR families.
Clinical Information of the Probands and Their Family Members
F and M in the “Age/sex” column represent female and male, respectively; A and UA in the last column in this table represent affected and unaffected, respectively.
Identification of causative mutations in four FEVR families
To identify the causative mutations in these FEVR families, WES was carried out to analyze the DNA samples from the 121 probands (overall WES results shown in Supplementary Table S1). After screening the mutations in known FEVR genes, we identified four novel mutations in the TSPAN12 gene, including c.1A>G (p.0), c.614G>A (p.G205D), c.695delT (p.V232Gfs*7), and c.833_842del (p.L278Qfs*25) may be responsible for the disease in four FEVR families, respectively. These mutations were verified by Sanger sequencing and were not found in our 2800 in-house WES database of non-FEVR controls. Further Sanger sequencing on another 500 ethnic-matched control individuals also did not find these mutations. Specifically, in family 1, we identified a heterozygous mutation c.1A>G in TSPAN12 in the proband II:2, and Sanger sequencing confirmed this mutation on his affected sister and mother, whereas his unaffected father did not carry this mutation (Fig. 1A, B). This mutation is an initial-lost mutation, and it led to untranslated or mistranslated TSPAN12 protein; thus, it is a pathogenic mutation. In family 2, a missense mutation in exon 8 of TSPAN12, c.614G>A (p.G205D) was identified to co-segregate with the phenotype (Fig. 1C, D). This mutation led to the replacement of Glycine by Aspartate. According to the genome Aggregation Database (gnomAD), which is provided on this website, it spans 123,136 exome sequences and 15,496 whole-genome sequences from unrelated individuals sequenced as part of various disease-specific and population genetic studies; the frequency of this mutation is very low (2/249512), implying its pathogenicity. As the TSPAN12 protein functions as an important co-receptor to enhance NORRIN/β-catenin signaling, it prompts us to test the protein activity harboring this missense mutation. To do this, we constructed a mutant TSPAN12 expression plasmid, and we proved that the expression of this mutant protein was comparable to that of the wild-type TSPAN12 plasmid (Fig. 3A). Further luciferase assays in HEK293STF cells indicated that the mutant TSPAN12 protein lost its activity to enhance the NORRIN/β-catenin signaling (Fig. 3B). These experiments proved that the p.G205D mutation was sufficient to impair the protein function, and thus was the pathogenic mutation for family 2. Through WES, we also identified two small-deletion mutations, c.695delT and c.833_842del in TSPAN12 gene from family 3 and family 4, respectively. These two mutations also co-segregated with the disease phenotype in the corresponding family (Fig. 2E-H).

In vitro functional studies on the G205D mutation TSPAN12 protein.
Discussion
FEVR is a complex inherited retinal disease with clinical and genetic heterogeneity, implicating the importance of molecular diagnosis. Mutations in the TSPAN12 gene have been reported to cause autosomal dominant FEVR (adFEVR). In this study, by using the WES method, we identified another four mutations in the TSPAN12 gene that cause adFEVR in four Chinese FEVR families. According to The Human Gene Mutation Database (HGMD), 25 mutations have been previously reported to cause FEVR, including 15 missense mutations, 5 splicing mutations, 4 small-deletion mutations, and 1 small insertion mutation. The identified four mutations in this study, c.1A>G, c.614G>A (p.G205D), c.695delT (p.V232Gfs*7), and c.833_842del (p.L278Qfs*25), were not included in the HGMD, and thus they were novel mutations to cause FEVR.
The TSPAN12 gene, located on human chromosome 7q31.31, encodes the 305 amino acid quadruple transmembrane protein TSPAN12 (Fig. 4), which is a member of the tetraspanin family (Maecker et al., 1997). TSPAN12 acts as a receptor of Norrin, binding with FZD4 and LRP5 to form a protein complex, which can enhance the signaling of Norrin but not Wnts. That is to say, when TSPAN12 combines with Norrin, it can further increase the Norrin/β-catenin signaling, whereas TSPAN12 does not have this effect when combined with Wnts (Junge et al., 2009). As shown in Figure 4, the four mutations were located in important domains of TSPAN12 proteins. Mutation c.1A>G (p.0) can lead to failed or uncorrected translation of TSPAN12 protein. Mutation c.614G>A (p.G205D), located at the large extracellular loop domain, which is located between the third and fourth transmembrane region (Fig. 4), is essential for combining with Norrin to enhance the signaling. This mutation may affect the binding of TSPAN12 to Norrin (Lai et al., 2017), and it may further impair the Norrin/β-catenin signaling. The last two mutations c.695delT (p.V232Gfs*7) and c.833_842del (p.L278Qfs*25) are located at the fourth transmembrane domain and C-terminal intracellular domain, respectively (Fig. 4), and they result in mistranslated TSPAN12 protein. According to Lai et al. (2017), changing the structure of transmembrane segments led to TSPAN12 losing the ability to enhance Norrin/β-catenin signaling, and TOP-Flash assays revealed that changing the structure of the intracellular C-terminus caused a partial reduction in signaling. Based on the facts just provided, the two mutations c.695delT (p.V232Gfs*7) and c.833_842del (p.L278Qfs*25) may strongly affect the TSPAN12 protein function, and thus are very likely to be pathogenic.
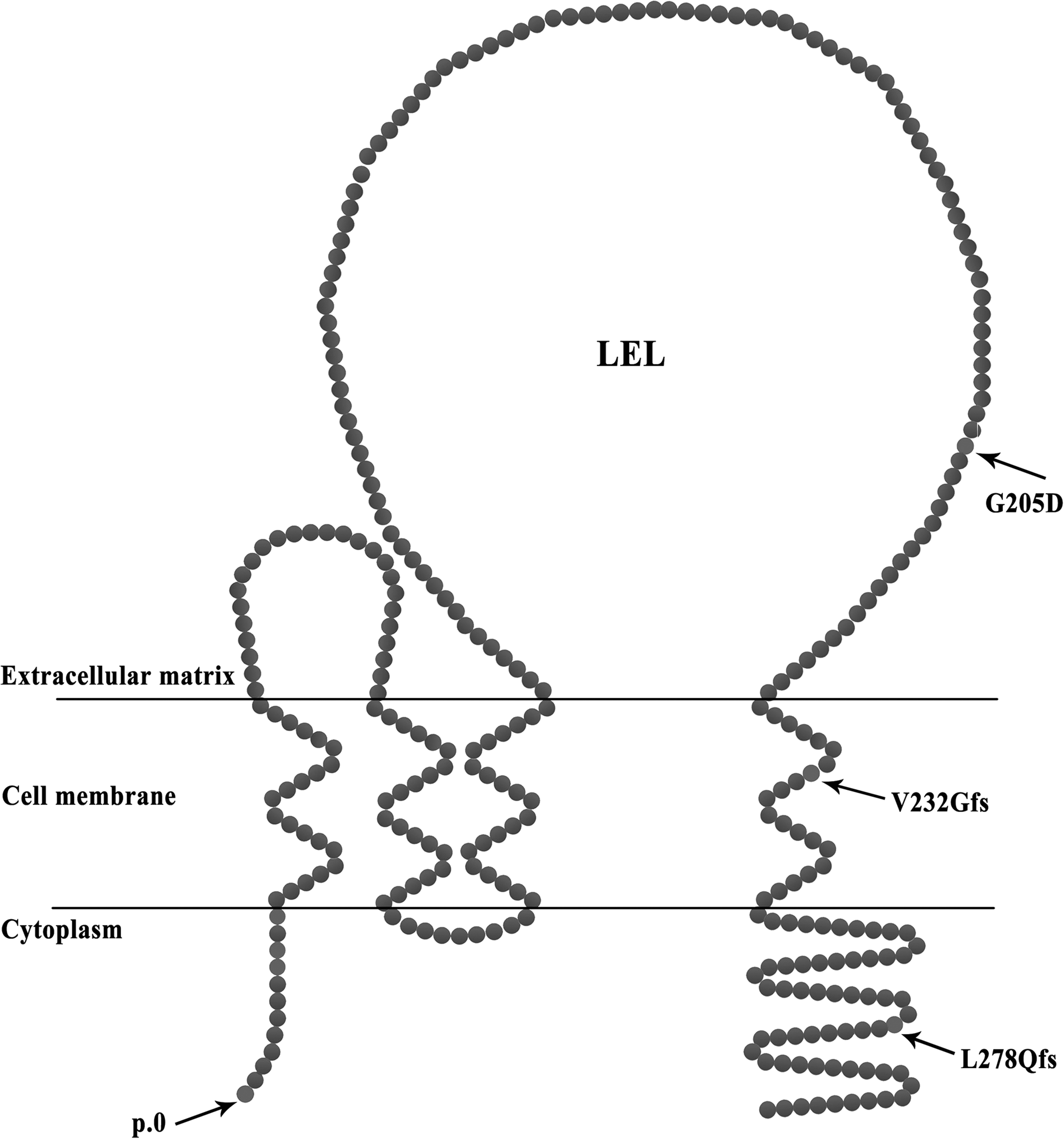
Schematic showing the TSPAN12 protein domains and the location of the mutations.
In conclusion, through WES analysis and functional studies, we identified four novel mutations in TSPAN12 gene in 121 Chinese FEVR families: c.1A>G (p.0), c.833_842del (p.L278Qfs*25), c.695delT (p.V232Gfs*7), and c.614G>A (p.G205D). These findings expanded the mutation spectrum of FEVR in the Chinese population, and they are thus of considerable diagnostic significance.
Footnotes
Acknowledgments
The authors thank all of the patients and their family members for their participation.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the National Natural Science Foundation of China (81700876 to L.Z.). It was also supported by grants from the Department of Science and Technology of Sichuan Province (2018YSZH0020 to L.Z.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.