
Abstract
Background:
Frontonasal dysplasia (FND) is a rare developmental disorder characterized by mild to severe changes in skull and brain structures. It is a phenotypically variable and heterogeneous disorder. This study was designed to provide a clinical and genetic analysis of FND in a consanguineous family of Pakistani origin.
Methodology and Results:
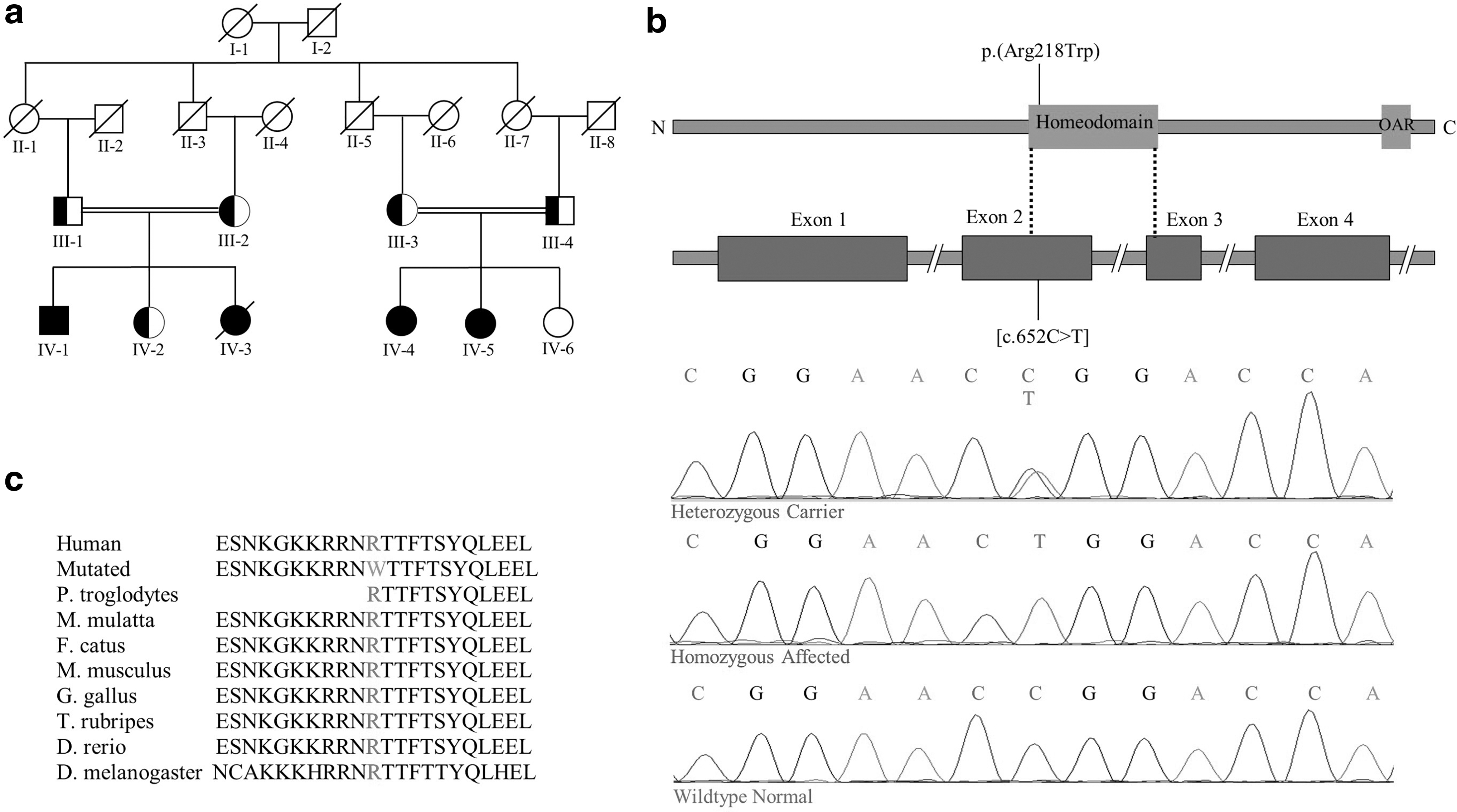
Affected individuals in the family showed characteristic features of frontonasal dysplasia type-2 (FND2), such as nasal bone hypoplasia, hypertelorism, and alopecia. Skull and brain imaging of affected members revealed ossification defects and various types of brain structural anomalies that created a split-brain. Sanger sequencing of the ALX4 gene revealed a homozygous missense variant [NM_021926.4: c.652C>T; p.(Arg218Trp)] in three affected members who demonstrated severe craniofacial anomalies. Heterozygous carriers in the family showed mild FND2 phenotypes.
Conclusion:
Clinical and genetic analysis of a family, exhibiting FND2 phenotypes, revealed several previously unreported clinical features and a novel missense variant in the ALX4 gene. These results will facilitate diagnosis and genetic counseling of the FND patients in the Pakistani population.
Introduction
Frontonasal dysplasia (FND) is a rare developmental disorder characterized by mild to severe changes in the structure of skull and brain. The condition has been classified into three types including FND1, FND2, and FND3. These forms are distinguished on the basis of underlying genes and distinct phenotypes. FND1 is distinguished from the other two forms by the appearance of wide philtrum with prominent philtral ridges, multiple bilateral swellings running into the nares, and medial depression of the nasal tip (Twigg et al., 2009). FND2 is characterized by missing nasal bone, completely flat nose with notched alai nasi, alopecia, parietal foramina (PFM), craniosynostosis, and brain malformation of posterior fossa (El-Ruby et al., 2018). Bilateral extreme microphthalmia, bilateral oblique facial cleft, and complete cleft palate are distinctive features of the FND3 (Uz et al., 2010). A mild phenotype of the FND3, which is likely a variant of the frontorhiny, has been reported as well (Ullah et al., 2017). FND1 and FND3 result from homozygous variants in the ALX3 and ALX1 genes, respectively (Twigg et al., 2009; Uz et al., 2010). However, FND2 develops from both homozygous and heterozygous variants in the ALX4 gene (Kayserili et al., 2012; Bertola et al., 2013; Altunoglu et al., 2014; Kariminejad et al., 2014).
In the present study, we have investigated a consanguineous family of Pakistani origin exhibiting characteristic features of FND2 such as nasal bone hypoplasia, hypertelorism, and alopecia. Sanger sequencing of the ALX4 gene revealed a novel homozygous missense variant p.(Arg218Trp) in affected members of the family. Heterozygous carriers in the family showed mild FND2 phenotypes.
Materials and Methods
This study was approved by the Institutional Review Board (IRB) of Quaid-i-Azam University Islamabad, Pakistan. Informed written consent, to conduct the study, was obtained from the family members. The patients were clinically examined at a government hospital. DNA of the family members was extracted from blood samples using QIAamp DNA Mini Kit [QIAGEN, Hilden, Germany].
Based on characteristic features of FND2, such as nasal bone hypoplasia, hypertelorism, and alopecia, observed in the affected individuals, a coding region of the ALX4 gene was sequenced in seven members (III-2, IV-1, IV-2, III-3, IV-4, IV-5, and IV-6) of the family (Fig. 1a). Considering the presence of PFM in the affected individuals, the coding region of the MSX2 gene was also sequenced. Purification of the polymerase chain reaction (PCR)-amplified exons of both ALX4 and MSX2 genes was carried out using QIAquick PCR purification kit [QIAGEN, Hilden, Germany]. The amplified products were Sanger sequenced at Eurofins Genomics [Eurofins, Germany]. The sequencing chromatograms were analyzed with BIOEDIT 6.0.7 sequence alignment software (Ibis Biosciences, Inc., Carlsbad, CA). Pathogenicity of the identified variant was assessed in silico using MutationTaster (Schwarz et al., 2014), PolyPhen-2 (Adzhubei et al., 2010), and SIFT (Sim et al., 2012). Frequency of the variant in the general population was searched in the gnomAD database.
Being a transcription factor, interaction of the ALX4 protein with DNA was assessed using wild-type and mutant homeodomains. The homeodomain structure of ALX4 [PDB ID: 2m0c (conformer# 0.1)] was retrieved from the Protein Data Bank whereas a palindromic DNA sequence (5′-AGAATAATCCGATTA-3′) was acquired from a previous study performed on ALX4 DNA binding (Tucker and Wisdom, 1999). HDOCK was used for DNA protein docking (Yan et al., 2017). A mutant ALX4 homeodomain was modeled on SWISS-MODEL server (Waterhouse et al., 2018). The possibility of the variant being present in nuclear localization signal (NLS) was assessed by cNLS Mapper (Kosugi et al., 2009). To assess role of the predicted NLS in nuclear transport, a model of ALX4 was generated by I-TASSER (Roy et al., 2010). The interaction of the modeled ALX4 with the nuclear transport proteins KPNA1 [PDB ID: 3tj3 (chain A)] was assessed by HDOCK. KPNA1 was selected as a putative import protein due to its expression in neurogenesis (Yasuhara et al., 2007) and ability to bind classical NLS sequences and homeodomain containing proteins (Ye et al., 2011). The protein structures and complexes were visualized by UCSF Chimera 1.13.1 (Pettersen et al., 2004). The interaction of ALX4 with KPNA1 was assessed through interface analysis by alanine scanning in mCSM-PPI2 (Rodrigues et al., 2019).
Results
Clinical description
Three affected members (IV-1, IV-4, and VI-5) of the family showed a severe form of FND2. The affected male member IV-1 had a developmental delay as he was unable to stand and walk at 2 years of age. He exhibited a prominent forehead, severe hypertelorism, upslanting palpebral fissure, sparse eyebrows, blepharophimosis, strabismus, broad and depressed nasal bridge, broad and depressed columella, cleft alae nasi, broad philtrum, and total alopecia (Fig. 2a). Two affected female members IV-4, aged 17 years, and IV-5, aged 19 years, exhibited similar severe facial features. All three affected family members had mild short stature, broad halluces, and short nail beds at noticeable distance from tip of the digits.

Clinical features observed in the family members by physical examination and CT scan of head and skull.
A head computed tomography (CT) scan of the affected member IV-1 revealed large ossification defects, coronal suture synostosis, bilateral foramina within the lamboidal sutures, and cranium bifidum like bilateral protuberances of soft tissue prominent on the prospective site of PFM (Fig. 2b-e). Brain imaging revealed lower insertion of tentorium cerebelli, relatively larger frontal lobes, absent splenium of corpus callosum, large suprasellar cistern with horns extended to the skull bones with wide fissures, and hypoplasia in the posterior region of the brain. Other prominent features present included cerebellar vermis hypoplasia, a deep interpedunclular fossa and bilateral clefts in the occipital lobe, wide lateral ventricles, and absent falx cerebri. A lateral invagination from the middle cortical region into the left ventricle developed into a deep sulcus that eventually gave rise to a fissure that along with a wide interhemispheric fissure created a split-brain appearance (Fig. 2f-o).
The individual IV-2, a carrier of the identified variant, had prominent forehead, short and sparse eyebrows, microphthalmia and underdeveloped right eye, anteverted nares, and mildly bifid nasal tip (Fig. 2p). She displayed two large connected PFM and various cranial vault findings (Fig. 2q-s). Brain imaging of the same individual IV-2 revealed cerebellar dysplasia, mild molar tooth sign, short lateral ventricles, and a number of other clinical features (Fig. 2t-y). Three heterozygous carriers (III-1, III-2, and III-3) in the family showed a variable combination of features including sparse eyebrows, short palpebral fissure, broad forehead, prominent eyebrow ridges, sharp bilateral edges of nasal ridge, mildly bifid nasal tip, and large bilateral PFM. One of the affected female babies (IV-3) was born with severe form of FND2 and died in infancy. The affected individuals in the family did not have any other systemic abnormalities.
Mutation analysis
Sanger sequencing of the ALX4 gene revealed a single nucleotide substitution [NM_021926.4: c.652C>T] in exon 2 resulting in a missense variant p.(Arg218Trp) located in the homeodomain (Fig. 1b). The variant was present in homozygous state in the three affected individuals (IV-1, IV-4, and IV-5) and heterozygous state in three other individuals (III-2, III-3, and IV-2). The member IV-6 had wild-type genotype. The amino acid Arg218 was found conserved across different species (Fig. 1c). The variant was absent from databases and predicted to have a damaging effect by MutationTaster, PolyPhen-2, and SIFT. Sequence analysis of the MSX2 gene did not reveal any variation.
Interaction of the wild-type and mutated ALX4 homeodomain with DNA revealed a decrease in binding energy for the mutated homeodomain (Fig. 3a-c). A monopartite NLS was predicted to lie within the sequence of 11 residues (211-KGKKRRNRTTF-221) with a maximum score of 7.5, suggesting a significant similarity to the classical NLS consensus sequence with a strong tendency for nuclear transport. The predicted NLS is located within the homeodomain of ALX4, which is similar to several other transcription factors containing NLS in the homeodomain (Ye et al., 2011). The NLS mapper also predicted a bipartite NLS sequence (211-KGKKRRNRTTFTSYQLEELEKVFQKTHYPD-240) within the homeodomain. A free docking of the modeled ALX4 protein with KPNA1 revealed involvement of the predicted NLS in the interaction (Fig. 3d). Interface analysis revealed involvement of the predicted bipartite NLS at 12 residues shown in capital letters (211-KgKKRRNrttftsyqleelekvfQKTHYPd-240) as predicted by the NLS mapper (Fig. 3e). The amino acid Arg218 did not appear on the interface and was located adjacent to the interacting residue Asn217.
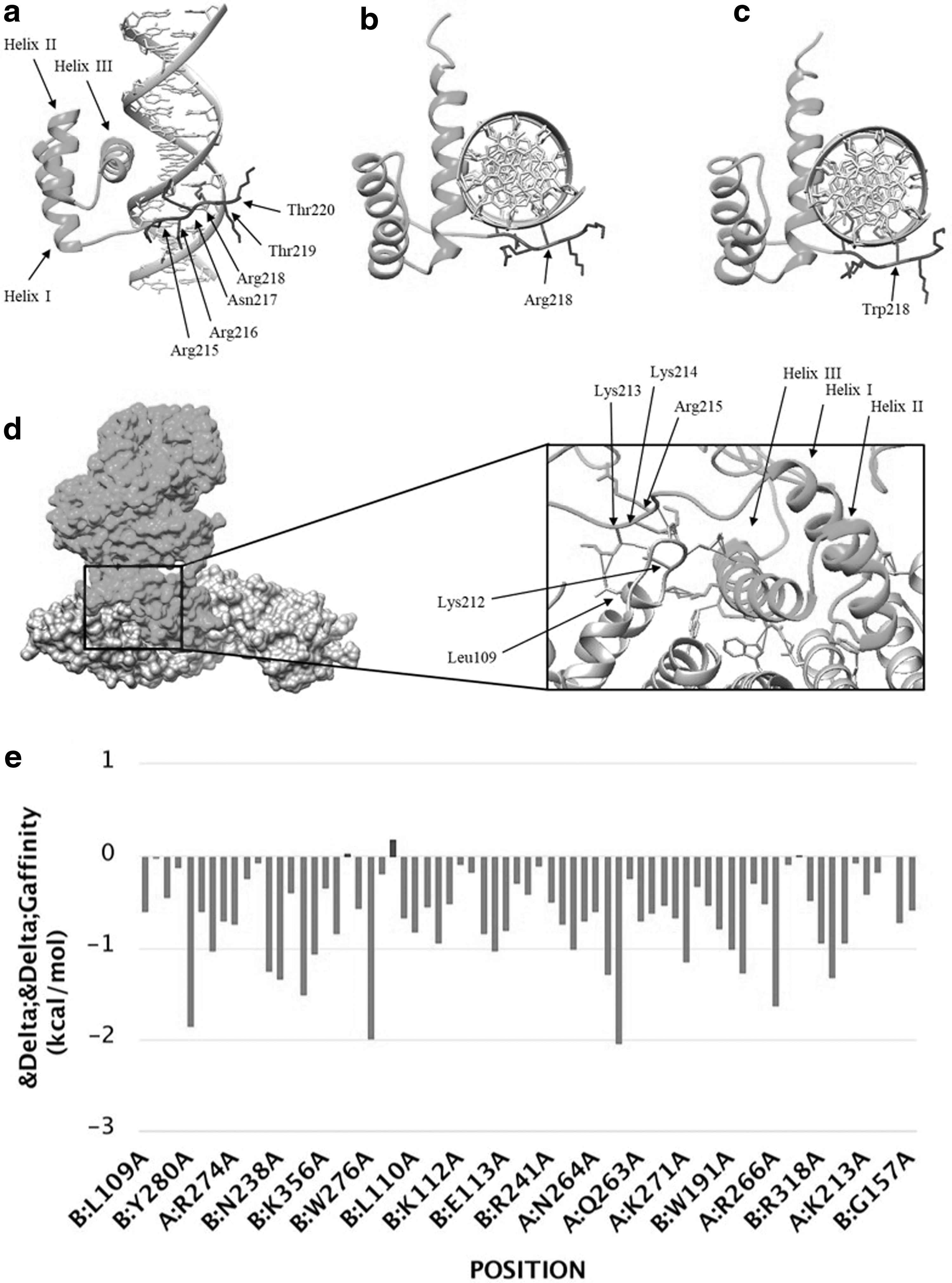
ALX4 docking to DNA. The HDOCK analysis showed top scores of binding affinity as −289.95 for wild-type (Arg218) and −284.32 for mutant (Trp218) homeodomain interactions.
Discussion
This study describes clinical and genetic analysis of a large consanguineous family segregating FND2 in an autosomal recessive manner. Screening of the ALX4 gene revealed a novel homozygous missense variant p.(Arg218Trp) in three affected members demonstrating severe form of FND2. The variant was found in heterozygous state in the carriers exhibiting mild frontonasal features.
The ALX4 gene, located on human chromosome 11p11.2, encodes 411 amino acid protein containing a homeodomain and an OAR domain and functions as a transcription factor (Qu et al., 1997). It is expressed in various organs and plays an essential role in the development of skull, limbs, and skin (Takahashi et al., 1998; Rice et al., 2003; Joshi et al., 2006). To date, about 28 variants in the ALX4 gene have been reported in causing various disorders, such as PFM, craniosynostosis, epilepsy, and FND2 (Supplementary Table S1). Among these, six homozygous mutations were involved in causing FND2 (Fig. 4). The variant p.(Arg218Trp), reported here, is the seventh homozygous variant leading to FND2.
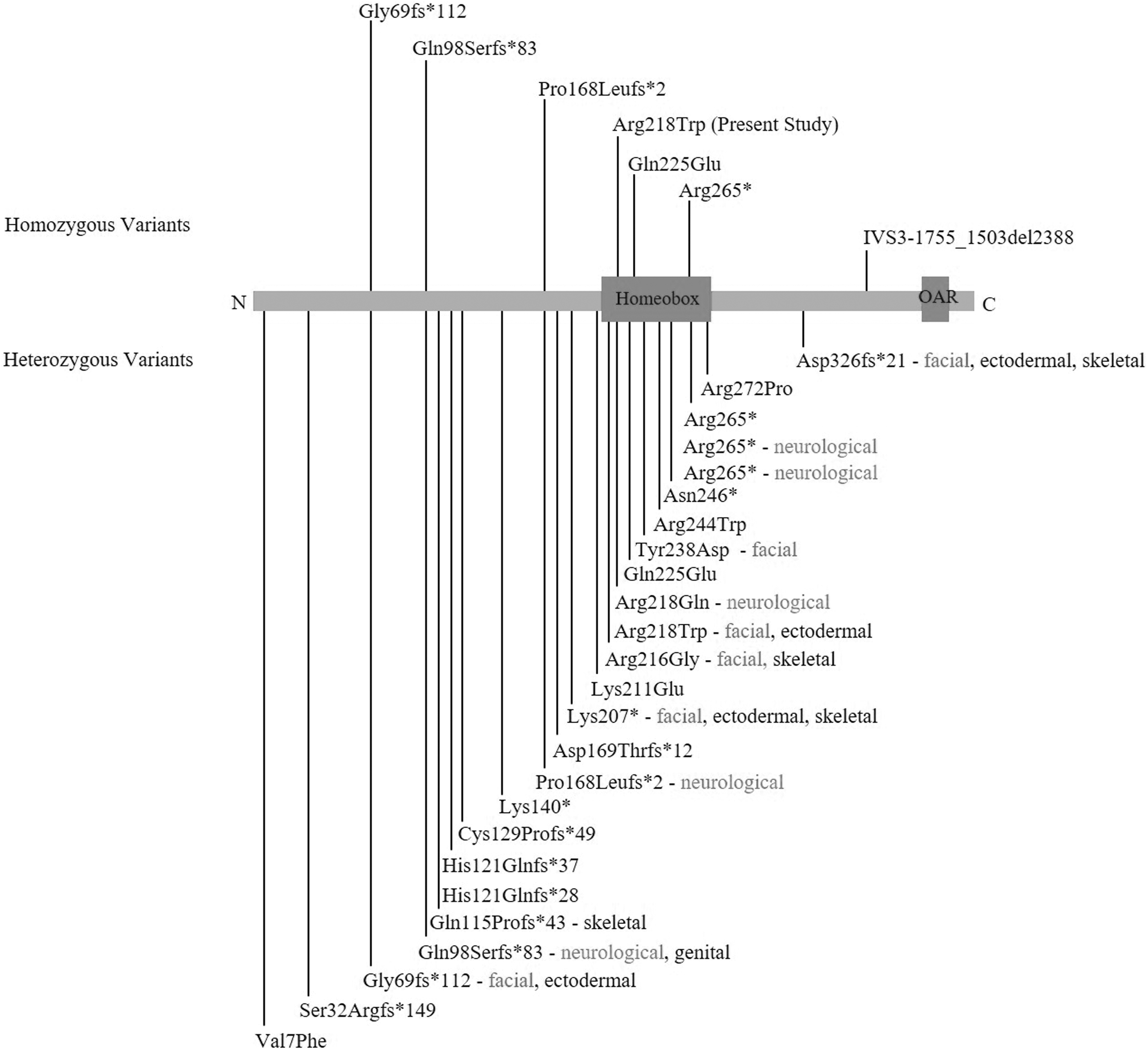
All heterozygous and homozygous coding variants positioned at the ALX4 protein with brief phenotypic categories. Almost all heterozygous variants with few exceptions are associated with PFM. In this diagram, other phenotypes are highlighted, detail and references of which are available in Supplementary Table S1 showing arrangement of all variants from N- to C-terminal of the protein.
In binding of the homeodomain containing proteins to DNA, basic amino acids, such as lysine and arginine at N- and C-terminals of homeodomain, play important role in stabilizing the interaction (Zucchelli et al., 2017). The variant in the ALX4 protein, identified in our family, is located at N-terminal region of the homeodomain and is predicted to result in reducing binding affinity for DNA (Fig. 3). In addition, it falls within the predicted NLS that has shown interaction with the nuclear transport protein KPNA1 (Fig. 3). Although, the residue Arg218 did not show interaction with KPNA1 in the predicted complex, but its role in binding importin cannot be excluded. Most likely this is due to limited accuracy of the in silico experiment in predicting structure of the ALX4 protein and availability of actual cytoplasmic environment in which the binding takes place.
The individual IV-1, carrying the variant in the homozygous state, showed numerous novel brain-related clinical features, including an abnormal fissure in the left cerebral region that along with agenesis of corpus callosum contributed to the split-brain appearance. The ALX4 protein is a genetic interactor of sonic hedgehog (SHH) that has been reported in causing holoprosencephaly and schizencephaly (Takahashi et al., 1998; Ribeiro et al., 2010). Development of the brain and limb abnormalities due to ALX4 disruption suggests involvement of the dysregulated No funding was received for this article. Majority of the heterozygous variants reported, to date, have been associated with PFM (Supplementary Table S1). Some of these variants lead to other features with no clear phenotype genotype correlation (Fig. 4). These different phenotypic manifestations may be distinct to a specific variant disrupting a unique set of ALX4 interactions with its partners. In addition, there might be an effect of a preexisting susceptibility background and de novo genetic modifiers that determine the type, severity, and penetrance of the phenotype. Large number of genes and regulatory elements participate in the developmental mechanisms including craniofacial morphogenesis (Attanasio et al., 2013; Brunskill et al., 2014). The severe underdevelopment of eye in the heterozygous carrier, IV-2, might be the effect of variants in those genes capable of modifying the ALX4-related feature of microphthalmia. The frontonasal phenotype resulting from both heterozygous and homozygous variants in the ALX4 gene is similar in severity in two previously published reports (Kayserili et al., 2012; Bertola et al., 2013), suggesting that FND2 follows both autosomal recessive and autosomal dominant mode of inheritance. In general, the recessive form is more severe and fully penetrant when compared with the dominant form, which sometimes shows incomplete penetrance as a result of dosage effect.
In conclusion, we have reported the first variant p.(Arg218Trp) in the ALX4 gene in a Pakistani family demonstrating variable FND2 phenotypes. The study will facilitate clinical diagnosis and genetic counseling of the FND patients in future.
Footnotes
Acknowledgments
Authors acknowledge and highly appreciate invaluable cooperation and participation of the family members in the study.
Website Resources
Protein Data Bank (https://www.rcsb.org/); SWISS-MODEL (https://swissmodel.expasy.org/); HDOCK (http://hdock.phys.hust.edu.cn/); NLS Mapper (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi); I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/); Chimera 1.13.1 (www.rbvi.ucsf.edu/chimera/); mCSM-PPI2 (![]() )
)
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.