
Abstract
Aims:
To explore the feasibility of detecting sex chromosome aneuploidies (SCAs) by means of gene copy number quantification of short stature homeobox (SHOX), vesicle-associated membrane protein 7 (VAMP7), and SRY in newborns.
Materials and Methods:
Gene doses of SHOX, VAMP7, and SRY were determined by quantitative polymerase chain reaction (qPCR) using DNA obtained from dried blood samples from newborns. Relative quantification values were obtained. An aneuploidy profile was established according to cutoff values. Samples with ≥2 gene doses (out of range) were reanalyzed, and those with aneuploidy profiles were confirmed by karyotyping. Sensitivity, specificity, and positive and negative predictive values were obtained.
Results:
A total of 10,033 samples were collected (4945 females and 5088 males). Of 244 (2.43%) samples with ≥2 gene doses that were retested, 20 cases were confirmed. The overall incidence of SCAs was 1 in 500 live newborns. There were six cases of Turner syndrome (1/824), 3 cases of XXX (1/1648), 7 cases of Klinefelter syndrome (1/726), and 4 cases of of XYY (1/1272). The sensitivity was 0.952 (95.42%); the specificity was 0.975 (97.56%); the positive predictive value was 0.909 (90.91%) and the negative predictive value was 0.987 (98.77%).
Conclusions:
Gene copy number analyses of the VAMP7, SHOX, and SRY genes by qPCR from blood samples spotted onto filter paper is a highly reliable method for the early detection of male and female SCAs.
Introduction
Sex chromosome aneuploidies (SCAs) are numerical anomalies of the X or Y chromosomes, which are the most frequent chromosomal aneuploidies in humans (Nielsen and Wohlert, 1990). Klinefelter syndrome (KS) (47,XXY) is the most common in men (1/500), and Turner syndrome (TS) (45,X), with a prevalence of 1/2500, is the most common monosomy (Nielsen and Wohlert, 1990). Such chromosomal abnormalities lead to an increased mortality and morbidity associated to a wide range of diseases (Berglund et al., 2019).
Clinical features of TS are short stature, webbed neck, and ovarian dysfunction, leading to infertility, osteoporosis, cardiovascular malformations, diabetes mellitus, and thyroid autoimmune diseases. Patients will benefit from early diagnosis since administration of growth hormone could give them the opportunity to reach a normal height and hormone replacement therapy; however, many girls with TS are not diagnosed until after 10 years old (Lee and Conway, 2014).
The clinical phenotype of KS patients is highly variable; the most relevant clinical characteristics are: average height 5-7 cm greater than a man of the same age and race without KS, hypergonadotrophic hypogonadism, and gynecomastia in about one third of individuals. Most patients have small testes and azoospermia, increased risk of breast cancer and endocrine and autoimmune diseases, and a 25% chance of developing osteoporosis (Grace, 2004; Gravholt et al., 2018). Hormone replacement therapy with testosterone should initiate at the beginning of puberty to reach adequate hormonal levels to initiate virilization, improve libido, osteoporosis prevention, and increase muscle mass. Infertility is a characteristic of KS patients; nevertheless, there are reports of patients with low fertility (Groth et al., 2013).
SCAs are not associated with obvious features at birth; therefore, only a small percentage of such disorders are detected in four newborns, for example, 10% for KS and 30-50% for TS (Stochholm et al., 2006; Herlihy et al., 2011). This may reduce the opportunity for early intervention, which may improve the quality of life of affected individuals (Lee and Conway, 2014).
There have been several efforts to detect SCAs using molecular technologies at different stages of life. Schmidt et al. (2000) reported a method for the prenatal diagnosis of somatic aneuploidies and SCAs based on the gene copy number. Other reports include the analysis by quantitative polymerase chain reaction (qPCR) of different genes for the diagnosis of TS, such as the androgen receptor (AR) gene (Rocha et al., 2010), dose-sensitive sex reversal-adrenal hypoplasia congenital critical region on the X chromosome gene 1 (DAX1), and the arylsulfatase E gene (ARSE) (Rocha et al., 2005), and for the diagnosis of KS, such as the AR and short stature homeobox (SHOX) genes (Ottesen et al., 2007; Aksglaede et al., 2012). We have previously published, in already diagnosed patients, that the identification of TS could be done by means of quantifying copy number of SHOX and VAMP7 (vesicle-associated membrane protein 7) pseudoautosomal genes together with the SRY (Ibarra-Ramirez et al., 2015).
The use of genetic markers for SCA diagnosis has been reported by several authors, either in already diagnosed patients or through next generation sequencing for prenatal diagnosis (Porreco et al., 2014; Zhang et al., 2017). In a previous report, Yoshimi et al. used fragile X-related epigenetic element 2, SRY, and FMR1 CGG repeat analysis, applied for fragile X syndrome screening in newborns, in the screening for SCA in 257 participants, aged between 19 and 69 years, in control males and females and SCA individuals, obtaining a high sensitivity and specificity (Inaba et al., 2013).
Several authors (Rocha et al., 2005; Watson et al., 2006; Rivkees et al., 2011) have reported high correlations in the diagnosis of SCAs with molecular techniques when comparing to the gold standard method (i.e., karyotyping). Nevertheless, these were performed in patients who already had a diagnosis of SCA. In another study, Correa et al., developed a method for neonatal detection of TS using ARSE and MAGEH1 gene dose quantification, which was tested in 996 newborns whose peripheral blood was collected and stored on filter paper, reporting a 95% detection sensitivity (Correa et al., 2014).
In the present study, we explore the feasibility of detecting SCAs by means of gene copy number quantification of SHOX, VAMP7, and SRY using dried blood samples from newborns spotted onto filter paper.
Materials and Methods
Patient data
A prospective blind study was performed at two hospitals in Nuevo León, México from May 2014 through July 2015. All live-born infants were eligible to participate, except for those that were transfused at birth. The protocol was approved by the Ethics Committee of the Facultad de Medicina y Hospital Universitario of the Universidad Autonoma de Nuevo Leon (registration no. GN13-002). Informed consent was obtained from parents or guardians, and a questionnaire addressing demographic data of the mother and neonates was completed for all participants.
Specimen collection
Blood samples were obtained by heel puncture in neonates at 24 h after birth, placed directly onto FTA Elute Micro Cards (no. WB120410; Whatman; GE Healthcare, Buckinghamshire, UK), and transported to the laboratory 2-3 days after collection. The cards were stored at room temperature in bags containing desiccants until further processing.
Inclusion criteria: only full term, apparently healthy neonates at 24 h postnatal life were included.
Exclusion criteria: presence of congenital defects or had received transfusions.
DNA extraction
DNA from FTA Elute cards was extracted according to the manufacturer's instructions, with the following modifications: three 1-mm disks from each sample were placed in a single well of a multiwell plate; 350 μL of Milli-Q water was added to each well and vortexed for 10 s. After a brief centrifugation, 45 μL containing the eluted DNA was transferred to new tube. Samples were incubated at 95°C for 20 min. The DNA from the samples was quantified using a NanoDrop 8000 spectrophotometer (Thermo Scientific, Wilmington, DE), and DNA concentration was adjusted to 10 ng/μL.
Determination of gene copy number by qPCR
Copy number quantification assays were performed using TaqMan probes (Applied Biosystems, Foster City, CA) following the manufacturer's instructions. The reference gene (RNaseP) was detected using a VIC-labeled TaqMan probe (catalog no. 4403328), whereas all other genes were detected using a 6-FAM-labeled TaqMan probe (Table 1). Real-time PCR was performed using a StepOnePlus Real-Time PCR System (Applied Biosystems), with 4 μL of 2 × TaqMan Genotyping Master Mix, 0.5 μL of each TaqMan probe (20 × ), 0.5 μL of the reference gene probe, and 5 μL of 20 × DNA to a final volume of 10 μL in a MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems). Amplification was performed under the following conditions: 95°C for 10 min, followed by 45 cycles at 95°C for 20 s, and 60°C for 1 min.
The Primers and Probes Used in This Study
SHOX, short stature homeobox; VAMP7, vesicle-associated membrane protein 7.
For normalization, the internal control gene RNaseP was amplified in parallel in the same well with the target gene under identical thermocycling conditions. The correlation coefficient between the cycle threshold (CT) and the DNA template concentration for each TaqMan assay was calculated by regression analysis. The amplification efficiency was calculated using the following formula: efficiency = −1 + 10(−1/slope).
Establishment of cutoff values
In this study, an analysis of global simulation based on the Monte Carlo method was designed and used. Considering the frequency of SCAs reported in several populations (1/400) (Nielsen and Wohlert, 1990), it was assumed that the probability of a positive sample is one in every five plates; therefore, every sample in each plate was used as a reference control for the other samples of each pool.
Briefly, data for each of the three target genes (SHOX, VAMP7, and SRY) were imported in plain-text format. For each batch, each sample was used in turn as a reference sample. Relative quantification (RQ) values were obtained using the delta-delta CT (ΔΔCT) method (Livak and Schmittgen, 2001). Each sample was classified as “loss,” “gain,” or “normal.” RQ ≥1.3 was considered a “gain,” whereas RQ ≤0.7 constituted a “loss,” and intermediate values were counted as “normal.” Those specimens with two or more genes with RQ values out of range (gain or loss) were reanalyzed in the same sample (second analysis, Fig. 1).

Study algorithm. A study algorithm was established to determine the profiles of SCAs according to the values obtained from the RQ of each of the analyzed genes (SHOX, VAMP7, and SRY). RQ, relative quantification; SCAs, sex chromosome aneuploidies; SHOX, short stature homeobox; VAMP7, vesicle-associated membrane protein 7.
An SCA profile was previously developed (Campos-Acevedo et al., 2016), and cases were classified as follows: in female neonates, when SHOX and VAMP7 RQ values were both above 1.3, possible XXX syndrome was indicated, and when the RQ values were below 0.7, this indicated TS (X monosomy). In male children, when SHOX and VAMP7 RQ values were above 1.3 with normal SRY, this was suggestive of KS (XXY), and when elevated RQ values of SHOX and VAMP7 were associated with increased values of SRY, XYY syndrome was suspected (Supplementary Data). Samples with results suggestive of an SCA after the second analysis were selected and the patients located for clinical evaluation and blood sampling to perform a karyotype to corroborate the diagnosis.
Quality control
Samples from patients with a confirmed diagnosis of SCA were included as positive controls. To identify false negative samples, a randomly selected subset of samples, from those that only showed one gene dose out of the normal range values, were retested. In addition, a subset from those that showed two or more genes out of range but without SCA profile was localized for karyotype.
Statistical analysis
The incidence of all SCAs and the incidence for each separate disorder were calculated.
The ability of the test (SCA profile established according to the reference cutoff values used) to identify true positives and negatives in a data set was measured by sensitivity and specificity as follows: sensitivity = TP/(TP + FN) and specificity = TN/(TN + FP) where FN was the false negative and FP was the false positive. MedCalc Statistical Software version 16.4.3 (MedCalc Software bvba, Ostend, Belgium, 2016) and IBM Corp. Released 2011. IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp.
Karyotype analysis
Peripheral venous blood (2-4 mL) from each newborn with a result that showed an aneuploidy profile was collected in a tube containing heparin for G banded karyotyping (30 metaphases) (Arsham et al., 2017).
Results
A total of 10,033 samples were analyzed, of which 4945 (49%) were from female newborns, and 5088 (51%) were male (distribution is shown in Fig. 2). From these, only 244 specimens (2.4%) had ≥2 gene doses out of range and were retested on the same sample, by triplicate. After retesting, 25 samples showed the SCA profile established (9 female and 16 male neonates); from these, only 22 patients accepted follow-up study and clinical evaluation. Twenty cases were confirmed as SCAs by karyotyping, and two resulted with normal karyotype and were considered false positive cases (Fig. 3).
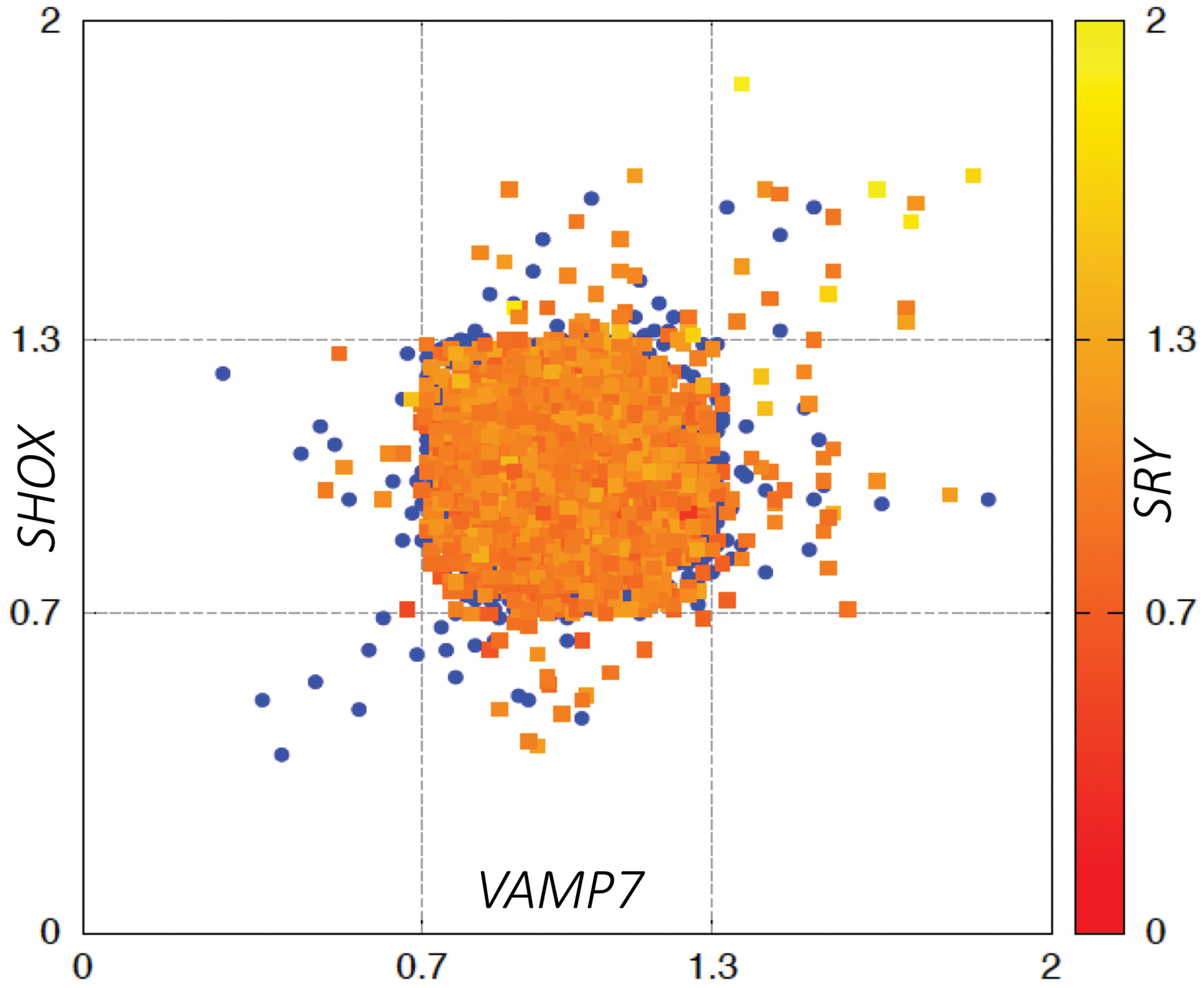
Scatter plot of RQ values of all samples, showing the distribution of female (blue circles) and male (squares) newborns, represented along the three axes according to the cutoff values. Samples that were out of range for the SRY gene are shown in red. Color images are available online.
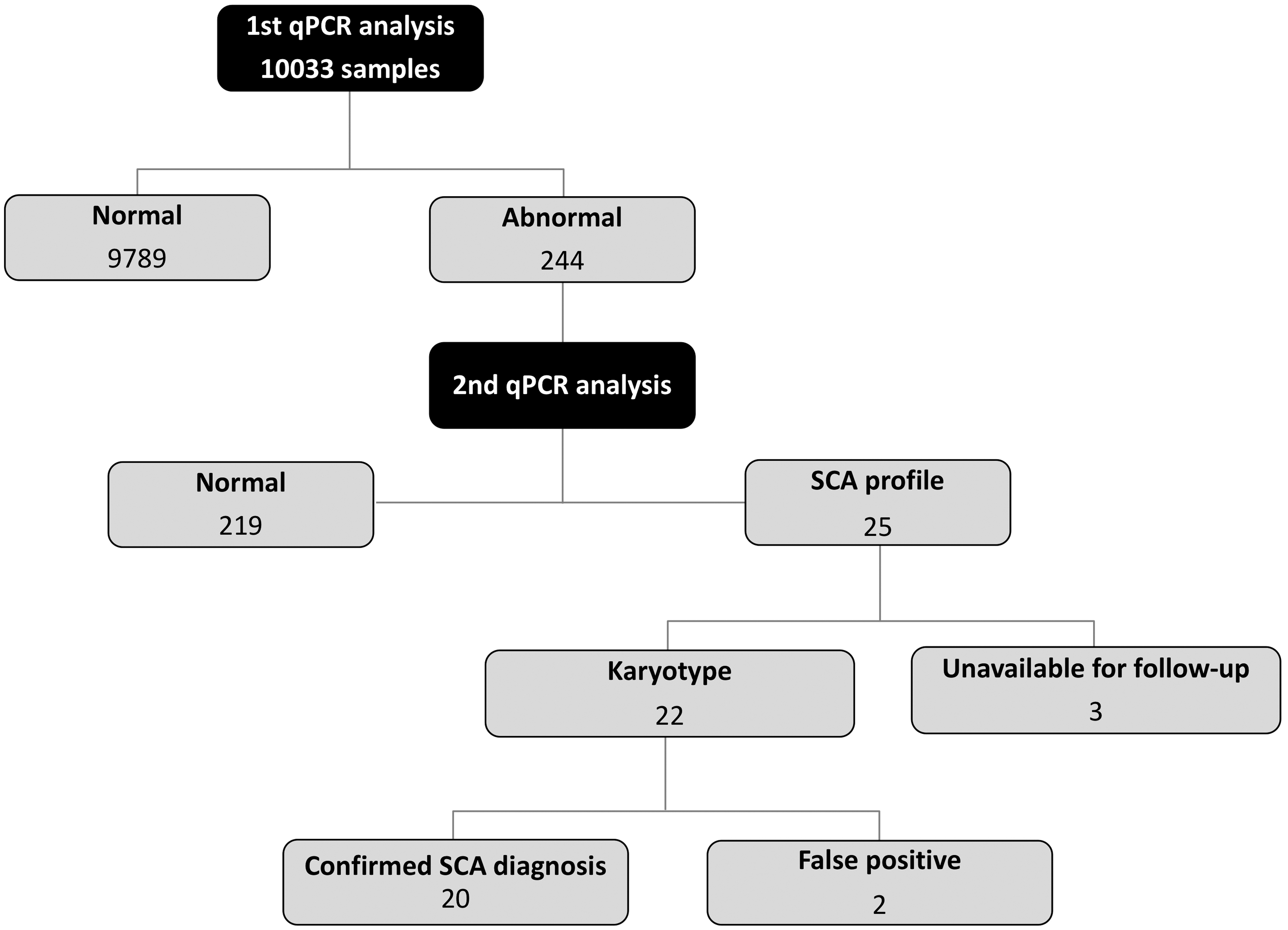
Result overview. The screening results of 10,033 newborns.
Table 2 shows the RQ values and karyotype reports. Nine female newborns resulted in abnormal karyotypes: 6 were confirmed as TS: 2 complete monosomies (45,X) and 4 mosaic: 45,X/47,XXX (1); 46 X, mar/46, XX (1); and 45,X/46,XX (2); the 3 other cases were X polysomies. From male samples, 16 showed an SCA profile, 9 were suggestive of KS, from which only 7 were confirmed by karyotype because 2 cases were lost in the follow-up; the other 7 samples showed an XYY syndrome profile, although confirmation was obtained only in 4 cases since in 2 cases karyotypes were normal (these were considered false positive) and, in one case, the parents refused to continue with the study.
Results from Patients Detected by Neonatal Screening. Relative Quantification Values of the Three Genes Analyzed by Quantitative Polymerase Chain Reaction Showing the Profile of Sex Chromosome Aneuploidies and G Karyotype Analysis Results from Patients Detected by Neonatal Screening
In one case, the parents refused to continue in the study (case 16), and two cases were lost to follow-up (cases 24 and 25); thus, karyotype analysis could not be performed.
Cases considered false positive (cases 14 and 15).
ND, no data; RQ, relative quantification.
One case with no SCA profile and RQ values of SHOX (0.789) and VAMP7 (0.794) in normal range but with an SRY (0.363) classified as loss gave us the suspicion of TS mosaicism. At clinical examination, the patient had a male phenotype, and the karyotype confirmed a mosaic of Turner (45,X/46,XY). In this case, a diagnosis of gonadal dysgenesis was established. This was considered as a false negative.
Quality control: All included positive controls showed a gene dose of VAMP7, SHOX, and SRY with an SCA profile that corroborated the reproducibility of the test by triplicate (data not shown). From those that only had one gene out of cutoff values (9789), a total of 439 samples were randomly chosen and retested, obtaining the same results. Eighty samples out of the 244 (32.7%), with abnormal RQ but not SCA profile after second test, were randomly selected and children localized for karyotyping, which all resulted as normal.
The overall frequency of SCAs was 1:500. TS was diagnosed in 1:824 female live newborns, and X polysomy was diagnosed in 1:1648 female live newborns, while KS and XYY syndromes were diagnosed in 1:726 and 1:1272 male live newborns, respectively. Sensitivity and specificity of the SCA test, using the established profile and the cutoff values obtained, were 95.24% and 97.56%, respectively, and positive predictive value (PPV) and negative predictive value (NPV) were 90.91% and 98.77%, respectively.
All newborns diagnosed with SCA had normal height and weight at birth. Only two patients with TS, with regular monosomy, had clinical data suggestive of the disease, such as heart defects (aortic coarctation and ventricular septal defect) and hyperpigmented macules. Two patients with XYY syndrome had micropenis and cryptorchidism. Nevertheless, only one case had been previously diagnosed before we did the evaluation and karyotype, and the others were diagnosed until they were summoned to our clinic for evaluation due to their abnormal screening test results.
Discussion
Several authors agree in the necessity of an early diagnosis of SCAs, particularly TS (Sarikaya, 2017) and KS (Herlihy and McLachlan, 2015), due to the increased knowledge about the treatment options that will benefit these patients. Even more, recently Berglun et al. (2019) reported that as SCAs are associated with increased morbidity and mortality and learning and/or behavioral disabilities, so the early diagnosis will provide better overall long-term outcome by providing an opportunity for timely intervention.
The pseudoautosomal regions (PAR1 and PAR2) are short regions of homology between the mammalian X and Y chromosomes. The PARs behave similar to an autosome and recombine during meiosis. In a previous study, we designed a methodology by means of quantification of gene copy number of SHOX (PAR1) and VAMP7 genes (PAR2) in both chromosomes and the SRY gene (only on the Y chromosome) to establish a molecular test for the diagnosis of SCAs (Porreco et al., 2014). This method was used in the present study allowing the identification of 20 patients with an SCA in male and female neonates (Turner, Klinefelter, XYY, and XXX syndromes).
The overall incidence of SCA cases found in this population from northeast Mexico was 1:500, which is similar to those reported in other populations, although the incidence of TS was 1:824, which is higher than that reported in the literature (1:2000-1:3000) (Nielsen and Wohlert, 1990). Contrary to previously reported data, where regular monosomy is the most common cause of this syndrome (Sybert and McCauley, 2004), the chromosome variant distribution of TS in our study showed that most patients were mosaics, and only 33% had monosomy.
However, these conflicting results could reflect the underdiagnosis of this disorder in patients with mosaicism because they show a less distinctive phenotype at birth (Koeberl et al., 1995). Our data are according to Tuke et al. who performed a study in adult women using single nucleotide polymorphism array technic and reported an overall prevalence of Turner of 1 in 1136 with a distribution of 1 in 8333 nonmosaic 45,X and 1 in 1315 for mosaic 45,X/46,XX, indicating that most mosaics are not detected maybe due to reduced penetrance. It is important to mention that with our method we detected other type of mosaics, which may explain the higher prevalence rate (Tuke et al., 2019).
We diagnosed three patients with X polysomy, which is one of the least diagnosed SCAs due to its variable phenotype (Otter et al., 2010; Tartaglia et al., 2010). The incidence of this disease in the present study was 1:1648, which was somewhat lower than reported previously (1:1000) (Nielsen and Wohlert, 1990).
KS was confirmed in seven patients, with an incidence of 1:726 newborn males, similar to that reported in the literature (Nielsen and Wohlert, 1990). These patients were not diagnosed at birth. KS is very difficult to diagnose because patients present only minor dysmorphias and normal height (Ratcliffe, 1999). Two cases with a KS profile in the newborn screening were lost in the follow-up and could not be confirmed by karyotyping; thus, these were not assigned as false positive or false negative.
The incidence observed in patients with XYY syndrome was 1:1272, which agrees with previously reported data (1:1000) (Nielsen and Wohlert, 1990). XYY syndrome and X polysomy are both underdiagnosed due to phenotype variability (Ratcliffe, 1999); nevertheless, some reports suggest that intellectual impairment and behavior problems are associated with these diseases (Cordeiro et al., 2012; Ulgiati et al., 2013); thus, earlier detection could provide an opportunity for better treatment of such complications (Lalatta et al., 2012).
In one case, a patient who did not show a SCA profile but with a low RQ value of SRY was clinically evaluated, and a karyotyping was performed. A diagnose of gonadal dysgenesis was done due to the male phenotype of the patient and the karyotype result (45,X/46,XY). In this case, it was possible to give a close follow-up and medical management. The incidence of this disorder is 1:15,000 (Muller et al., 1999) in newborns; in the present study, we found 1 case in 10,000 newborns. Nevertheless, this case was considered false negative. Two cases that initially showed the XYY profile revealed a normal karyotype. These were considered as false positive cases.
For the diagnosis of SCA, the correlation of the cases detected with the method presented here with karyotyping was 90%. Since only 10-30% of SCAs are detected at birth (Stochholm et al., 2006; Herlihy et al., 2011), the detection rate of these pathologies in newborns with our methodology will significantly increase, allowing the opportunity for prompt and adequate intervention. Sensitivity and specificity of the SCA profile were 95.24% and 97.56%, respectively, and PPV (90.91%) and NPV (98.77%).
Compared to other technics such as multiplex ligation-dependent probe amplification (MLPA), our method is less expensive and less time consuming; besides, MLPA is not suitable for use in high throughput dry blood spot screening method. The methodology used in the present is an important tool because it is quick and easy to screen for sex chromosome aneuploidies, as the karyotype is time consuming and it can only be performed in specialized laboratories of cytogenetics.
To our knowledge, this is the first large-scale study that used a molecular approach for the early detection of SCA, which is capable of detecting mosaics.
Conclusions
Gene copy number analysis of VAMP7, SHOX, and SRY genes by qPCR in blood samples spotted onto filter paper is a good method for the early detection of male and female SCAs.
Footnotes
Acknowledgments
The authors thank the volunteers and staff from the Hospital Universitario José E. González, Universidad Autónoma de Nuevo León (UANL), and Hospital Regional Materno Infantil from the Nuevo Leon Health Services, as well as the personnel of the Department of Genetics from the School of Medicine (UANL).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT) CLAVE SALUD (2013-1-202628).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.