
Abstract
Aims:
Identification of genetic mutations linked to hereditary multiple osteochondromas (HMO) is crucial for understanding the molecular mechanisms leading to disease pathogenesis. In this study, we investigated four patients and eight healthy individuals from a family with HMO.
Methods:
Clinical HMO data and Sanger sequences of the coding regions of the exostosin glycosyltransferase 1 (EXT1) gene (18q24.11) and the EXT2 gene (11p12) of all 12 members of the family were analyzed.
Results:
A novel nonsense mutation in the EXT2 gene (c.526C>T; p.Gln176*) was detected, which was present in all four patients but absent in their healthy relatives. This mutation encodes a stop codon that results in a truncated EXT2 protein that consists of only 176 amino acids and lacks the remaining 522 amino acids at its C-terminus, missing the entire glycosyltransferase domain.
Conclusions:
Association of a truncated EXT2 protein with HMO provides new insights into exostosis pathogenesis, highlighting potential roles of the EXT2 gene and its glycosyltransferase domain. Further research is required to understand the mechanisms underlying the development of exostosis.
Introduction
Hereditary multiple osteochondromas (HMO) is a hereditary autosomal dominant disease, characterized by multiple benign tumors mostly located at the epiphysis of long bones under cartilage caps osteochondromas causes skeletal deformity, short stature, and joint dislocation, which affect joint movement, compress the surrounding tissues, and can even lead to malignant transformation. These pathologies may result in limb disability and mental disorders. Surgical resection is performed when osteochondromas result in the aforementioned severe phenotypes.
Current studies suggest that the HMO is associated with at least three genes: exostosin glycosyltransferase 1 (EXT1) (Cook et al., 1993) located at 8q24.11-q24.13, EXT2 (Wu et al., 1994) at 11p12-11, and EXT3 (Le et al., 1994) at 19p. EXT1 and EXT2 have been cloned (Ahn et al., 1995; Stickens et al., 1996; Van Hul et al., 1998), but it is not possible to distinguish clinically which gene mutation is responsible for the disease. The phenotypes of HMO are various, and there may be other gene mutations with modifying effects. Therefore, it is speculated that the EXTL gene family that has the same carboxyl end coding product as the known EXT gene is also likely to be a candidate pathogenic gene of HMO (Hall et al., 2002). It includes three members in EXTL gene family: EXTL1 at 1p36.1 (Wise et al., 1997), EXTL2 at 1p11-p12 (Wuyts et al., 1997), and EXTL3 at 8p12-p22 (Van Hul et al., 1998). But so far, no EXTL mutation has been reported in HMO patients. In physiological environment, normal levels of heparan sulfate (HS) and EXT expression maintain the intrinsic mesenchymal phenotype of chondrocytes and block signal transduction through locally expressed bone morphogenetic protein (BMP) (Anower-E-Khuda et al., 2013). Mutations in EXT1 or EXT2 result in about 50% systemic HS deficiency (Bandyopadhyay et al., 2008). Decreases in local HS levels disrupt the signaling pathway for maintaining the cartilage membrane stroma, lead to BMP overexpression, and cause formation of heterotopic cartilage and osteochondromas (Pacifici, 2018).
In this study, we analyzed the genomes of a family (three generations) with HMO by polymerase chain reaction (PCR) followed by Sanger sequencing, and identified a novel heterozygous nonsense mutation in EXT2 (c.526C>T; Gln176*), which can be used for prenatal genetic diagnosis and facilitate further studies on HMO pathogenesis.
Materials and Methods
Subjects and clinical evaluation
Patients included in the study were from a Han Chinese family from Hangzhou, China. The family had 12 members belonging to three generations, including 4 HMO patients. All four HMO patients were male. The oldest patient was 81 years old, whereas the youngest was 8 years old. The proband, 42 years old, was admitted to our hospital in February 2017 because of a protruding joint mass around his knees, but he refused to undergo surgery. All family members agreed to participate in the study and signed informed consent forms. Our study was approved by the Tongde Hospital of Zhejiang Provincial Ethics Committee. All individuals in the family were investigated in detail. X-rays and other auxiliary examination data of some patients were collected to establish the family clinical database. The data included age of onset, age of diagnosis, major symptoms and discomfort, distribution, size and shape of osteochondromas, history of surgery, tendency to malignancy, neurological symptoms, and cause of death. The peripheral venous blood samples of the proband, his father, siblings, and children were collected (5 mL/person) and stored in vacuum anticoagulation vessel at −20°C until analyses.
Mutation screening
Next, the DNA was purified from blood samples using AxyPrep Nucleic Acid Purification kit (Axygen Scientific, Inc., Union City, CA) according to the manufacturer's protocol. DNA concentration was measured using a NanoDrop 2000 spectrophotometer (Nanodrop Technologies; Thermo Fisher Scientific, Inc., Wilmington, DE). Primer 5.0 (Premier Biosoft International, Palo Alto, CA) was utilized to design primers (Table 1) for all exons of EXT1 and EXT2 (including the 100-bp region of exon and intron junctions). We performed PCR (KAPA2G Fast Multiplex Mix; Kapa Biosystems, Inc., Wilmington, MA) to expand genomic DNA with the following cycling conditions: 30 cycles of 5 min at 94°C, 30 s at 94°C, and 30 s at 60°C; followed by 1 min at 72°C, 5 min at 72°C, and 10 min at 4°C. Sanger sequencing of amplified DNA product was performed using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA) and an ABI 3730XL automatic sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.). Samples from the patients and healthy controls were bidirectionally sequenced and the sequencing results were analyzed with Sequencer Demo 3.0 (Gene Codes Corporation, Ann Arbor, MI) and Mutation Surveyor Demo V4.0 (SoftGenetics, LLC, State College, PA). Gene sequences obtained from National Centre for Biotechnology Information (EXT1, NM_000127.2; EXT2, NM_001178083.1) were used as reference.
Primers for All Exons of EXT2
EXT, exostosin glycosyltransferase.
Results
Drawing pedigrees and clinical manifestations
We investigated a three-generation family consisting of 12 individuals, among whom 4 exhibited HMO with autosomal dominant inheritance (Table 2). The morbidity rate was 33.3% and all affected individuals were males. The proband was 42 years old, whereas the other three patients were 8, 52, and 81 years old. The average adult height was 164.5 cm for males and 155.6 cm for females (no short stature). In all patients, the age of onset for osteochondromas growth was before 10 years. None of the subjects showed a tendency for malignant transformation and none of the patients received surgical treatment. The members of the family are shown in Figure 1.

A family with HMO in Chunan, Hangzhou. The proband (II5; fifth member of the second generation of the family) is marked with an arrow. The filled squares represent affected individuals, and empty circles and squares indicate unaffected family members. There is no relationship by consanguinity in this family. Squares indicate male patients; circles indicate female patients. HMO, hereditary multiple osteochondromas.
Clinical Data for Four Hereditary Multiple Osteochondromas Patients
When the proband was 8 years old, an abnormal mass around the knee joints was prominent and gradually enlarged until he was 22 years old. Different sizes of tumors were detected by examination in right ilium, right hip joint, and around knee joints. The biggest tumor mass was about the size of a walnut, and the smallest was about the size of a peanut. The tumors were hard in texture, and they did not move upon pushing or with joint movement. There was no local fluctuation, pulsation, obvious tenderness, or limitation of joint function. X-ray images (Fig. 2) showed multiple osteochondromas (MO) in the lower femur, upper tibia, and fibula. The proband's mother and her family had no relevant medical history, whereas his father (I1), son (III3), and brother (II3) had MO throughout the body with mild symptoms.
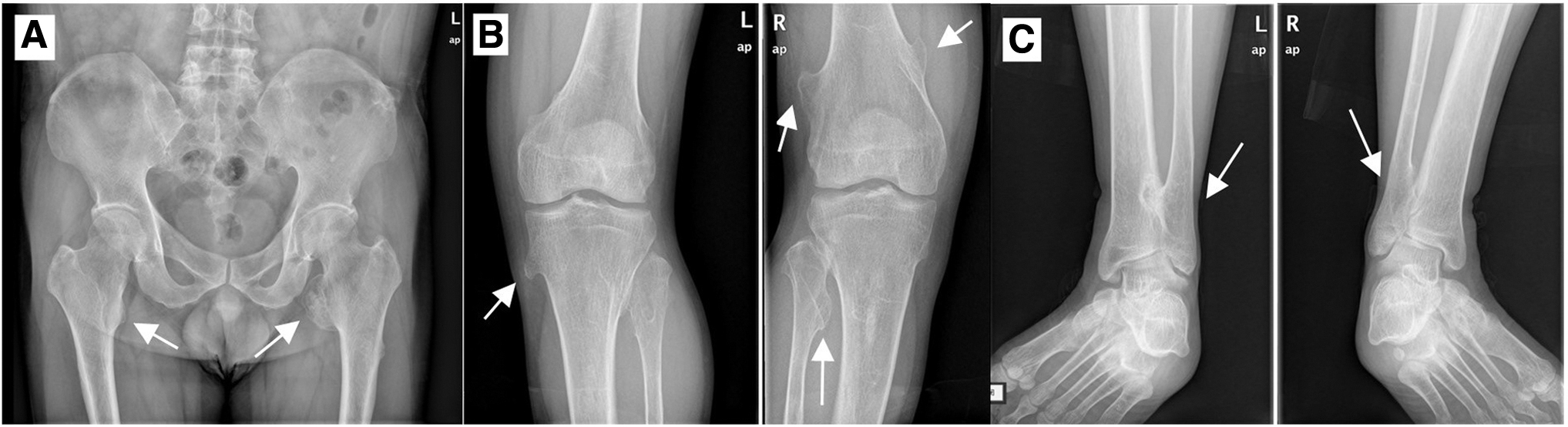
Imaging examination of HMO probands.
Sequencing results
By sequencing four patients and eight healthy relatives, we identified a nonsense mutation in exon 2 of EXT2 gene in the proband and other patients. This mutation (c.526C>T; p.Gln176*) was not detected in other family members or healthy controls (Fig. 3A). It was confirmed through a search on the Human Gene Mutation Database (HGMD®) that the mutation had not been reported to date, and that the mutation locus is a new mutation site. We also found two synonymous mutations in exon 9 of EXT1 (c.1761G>GA; p.587E>E/E), and in exon 2 of EXT2 (c.127C>A; p.43R>R).
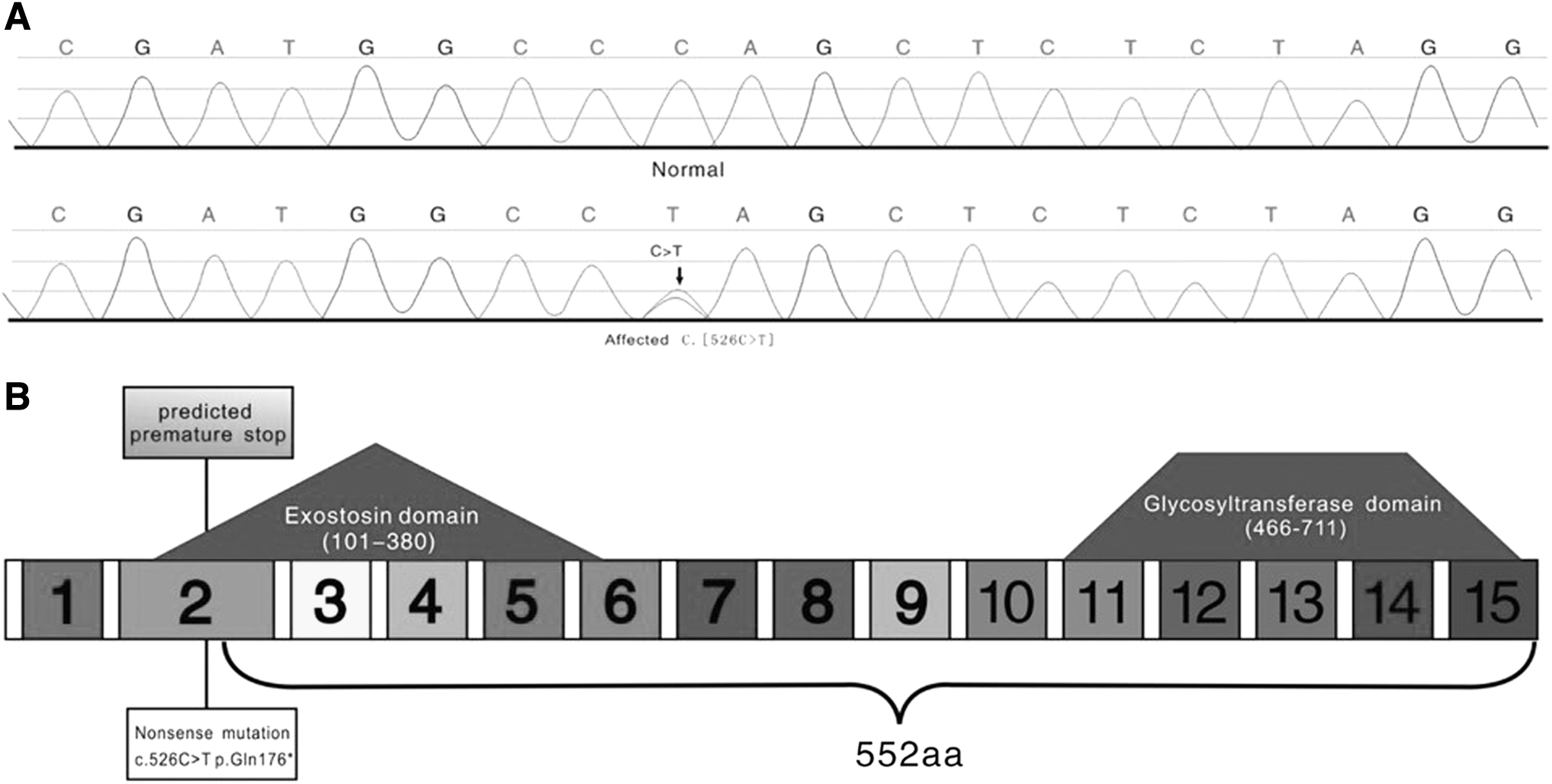
EXT2 mutation.
Computer simulation analysis
We used computer simulation analysis to further understand the effect of Gln176* mutation on EXT2 gene function. The 728-amino acid protein encoded by EXT2 gene had two important domains: an exostosin domain (amino acids 101-380) and a glycosyltransferase domain (amino acids 466-711) (Fig. 3B). Moreover, evolutionary conservation analysis revealed that amino acid 176 (glutamine) located in exostosin domain was highly conserved among species, suggesting that it has an important role in the function of EXT2 gene. The nonsense mutation Gln176* results in premature termination of transcription and produces a truncated EXT2 protein that lacks the last 552 amino acids at its C-terminal (Figs. 3B and 4). Glycosyltransferase domain, which is located at the C-terminal of EXT2, plays an important role in HS biosynthesis. Therefore, the nonsense mutation Gln176* may have a significant impact on the function of EXT2 gene, especially on HS biosynthesis. We confirmed this prediction with the online tool Mutation Taster, which showed that substitution of amino acid 176 by a termination codon would probably lead to the EXT2 dysfunction and disease (probability score, six points).
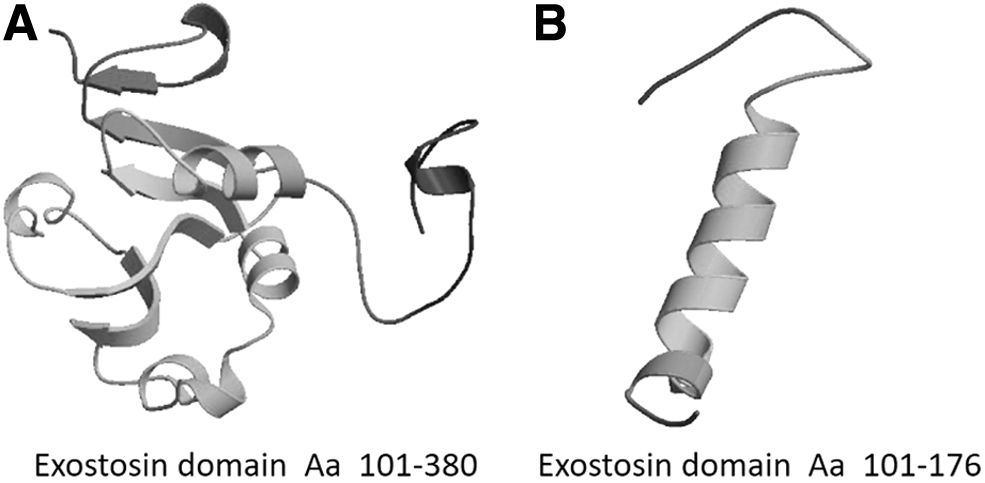
The comparison between exostosin domains of mutant (c.526C>T; p.Gln176*) and wild type EXT2s.
Discussion
Osteochondromas are mostly solitary and nonhereditary. Although a large majority of patients exhibit a single osteochondroma (Koehl and Tilson, 1977; Volpi et al., 1986), about 15% of patients would have two or more osteochondromas, called MO (Hennekam, 1991; Paik et al., 2000). Most of these patients (>70%) are diagnosed with HMO that runs in families. Osteochondromas usually appear at the early stages of growth and development. However, it is rarely diagnosed early because the chondroma body needs to reach a considerable size before symptoms can occur. More than 85% of patients with HMO have mutations in EXT1 or EXT2. Among those, 78% have mutations in EXT1 and 21-44% in EXT2 (Cardelia et al., 1995; Porter and Simpson, 1999; Dobson-Stone et al., 2000; Francannet et al., 2001; Gigante et al., 2001; Hall et al., 2002; Vink et al., 2005; Lonie et al., 2006; Jennes et al., 2008). In contrast, mutations in EXT2 are more common than those in EXT1 in Chinese pedigrees (Xu et al., 1999). Some studies also suggest that the HMO phenotype associated with EXT1 mutations seems to be more severe than that caused by EXT2 mutations (Porter et al., 2004; Alvarez et al., 2006, 2007; Jager et al., 2007). Generally speaking, the mutation of EXT2 gene causes a relatively mild phenotype. Clinical symptoms of male patients are more advanced than those of female patients, which may be related to the later closure of growth plate in male patients and the longer time allowed for the growth of osteochondromas; patients with a larger number of osteochondromas carry a higher risk of deformity or disability (Francannet et al., 2001; Alvarez et al., 2006; Pedrini et al., 2011).
Previously, we found a mutation in EXT1 (c.1902C>A; p.Tyr634*) (Cao et al., 2014) and two mutations in EXT2 (c.67C>T; p.Arg23* and c.660delG; L221Cfs*82) (Chen et al., 2018; Ruan et al., 2018) associated with osteochondromas. In this study, members of a Chinese family with osteochondromas were investigated, and a novel nonsense mutation (c.526C>T p.Gln176*) as well as a synonymous mutations (c.127C>A; p.43R>R) in EXT2 were identified. All four patients in this family exhibited abnormal bone growth before they reached 10 years of age. They had osteochondromas on both hips, knees, and ankles, but these lesions neither transformed into malignancy nor exhibited compression of blood vessels and nerves.
Clinical manifestations of MO vary even among patients from the same family. At present, no characteristic set of symptoms has been associated with a particular mutation; the pathological and biochemical mechanisms of multiple osteochondroma are not clear. Studies have shown that the proteins encoded by EXT1/2 genes are transmembrane glycoproteins located in the endoplasmic reticulum. They show glycosyltransferase activity and also participate in the synthesis of heparan sulfate proteoglycans (HSPGs) in vivo. HSPGs play an important role in the regulation of various cell signaling molecules. Overall, it is thought that gene mutations induce defects in signaling pathways (India hedgehog, fibroblast growth factors, and bone morphogenic proteins) leading to differentiation of some perichondral progenitor cells into chondrocytes. However, these chondrocytes are not arranged longitudinally, instead, they grow perpendicular to the growth plate and gradually form osteochondroma (Ryckx et al., 2013). Further research is required to pinpoint the roles of EXT genes in these signaling pathways and osteochondroma pathogenesis.
Summary
This study confirmed that the nonsense mutation in EXT2 gene (c.526C>T; p.Gln176*) is the molecular mechanism of HMO in this family. The result can be used for genetic consultation and prenatal gene diagnosis of the family members and provides basis for further study of the molecular mechanism of HMO.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (grant no. 81672769).