
Abstract
Objective:
To evaluate the prognostic value of driver mutations in the KRAS, CDKN2A/P16, TP53, and SMAD4 genes in pancreatic cancer to aid in the design of therapeutic strategies.
Search Strategy:
A systematic search was conducted using PubMed, MEDLINE, Springer, and Cochrane library to identify eligible studies published between January 1990 and June 2018 that reported an association between driver mutations in these genes and survival data.
Inclusion Criteria:
Articles which passed the primary screen were further scrutinized for the presence of all the following items: (1) cohort studies or case-control studies, evaluating the relationship between driver mutations and cancer; (2) cancer diagnoses clearly proved; and (3) hazard ratios (HR) and 95% confidence intervals (CIs) were characterized by sufficient information.
Data Extraction and Analysis:
Selection of included articles, data extraction, and methodological quality assessments were, respectively, conducted by two authors.
Results:
The meta-analysis was composed of 17 studies on the P53, 8 on SMAD4, 7 on CDKN2A/P16, and 2 on KRAS, containing 3373 samples. Our pooled results demonstrated that the patients with overexpression of the P53 (HR = 1.249, 95% CI = 1.003-1.554, p = 0.047), SMAD4 (HR = 1.397, 95% CI = 1.015-1.922, p = 0.040), CDKN2A/P16 (HR = 0.916, 95% CI = 0.583-1.439, p = 0.704), and KRAS (HR = 1.68, 95% CI = 1.27-2.22, p < 0.001) mutations all had poorer overall survival.
Conclusion:
This systematic review and meta-analysis supports the use of driver mutations in the P53, SMAD4, and KRAS genes as prognostic markers for pancreatic cancer.
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the United States and Europe (Del Chiaro et al., 2017; Moutinho-Ribeiro et al., 2017; Soldan, 2017). It is essential for us to be aware of this, one of the most fatal diseases, which is due to its late stages of diagnosis, aggressive phenotypes, and resistance to current therapies. Only a small part of patients had a diagnosis at a stage at which they still benefited from tumor resection. Surgery remains the primary treatment strategy and the only chance of cure for pancreatic cancer. Furthermore, the 5-year survival rate of such postoperative patients was reported to be 9% to 21% (van Rijssen et al., 2016; Buanes, 2017; Strobel et al., 2017). Unfortunately, some of such resectable patients suffered from rapid demise and poor prognosis after surgery, suggesting that these patients diagnosed as a resectable disease, in fact, had distant occult metastases. Sadly, nonsurgical treatment modalities for this malignancy have all too often yielded minimal results (Guo et al., 2013). In contrast to other malignancies, such as breast and lung cancer, where the incorporation of molecular assessment has become part of routine practice for therapeutic stratification, current treatment algorithms for pancreatic cancer still depend on only imaging and histological assessments to guide treatment and help classify prognosis (Fisher et al., 2017). It is beneficial to stratify patients and to develop more individual therapies (Wirth and Schneider, 2016).
The poor response to current therapies in pancreatic cancer results from biological aggressiveness, which is caused by multiple genetic variations in cancer cells within the tumor mass (Cicenas et al., 2017). Recent studies showed that the growth and development of pancreatic cancer involved some genetic variations in oncogene activation, loss of tumor-suppressor gene function, and overexpression of the receptor-ligand system (Jones et al., 2008; Oshima et al., 2013). Four major genes, including KRAS, TP53, CDKN2A/P16, and SMAD4, are the most frequently mutated genes in the landscape of the human pancreatic ductal adenocarcinoma (PDAC) genome as revealed by whole-exome sequencing (Jones et al., 2008). Mutations in all four genes are recognized as “driver mutations” in pancreatic cancer because they drove neoplastic transformation (KRAS) and tumor progression (CDKN2A/P16, TP53, and SMAD4) (Kamisawa et al., 2016). The concept that these four genetic mutations are indeed driver mutations has been proved in several studies (Cowley et al., 2013; Dickson, 2017; Hayashi et al., 2017). Recent studies reported that targeted pancreatic activation of KRAS from its endogenous locus results in pancreatic intraepithelial neoplasia (PanIN) lesions that advance to pancreatic cancer after a long latency and that concomitant deletion or mutation of CDKN2A/P16, P53, or SMAD4 cooperate to produce metastatic pancreatic ductal adenocarcinoma (Hingorani et al., 2003, 2005; Korc, 2010).
Oncogenic KRAS mutation is a frequently observed hallmark of a variety of human cancers, and its role in pancreatic cancer initiation and progression is well described. Mutational activation of the KRAS oncogene has been reported in 47% to 100% of pancreatic cancer (Kamerkar et al., 2017). In the current mouse model of pancreatic carcinogenesis, oncogenes KRAS mutations in codon 12 led the development of precursor lesions (pancreatic) that progress spontaneously to pancreatic cancer. Therefore, the mutation of the KRAS gene seems to be closely related to cancer initiation (Avila et al., 2012; Fendrich et al., 2013). However, it is still obscure that KRAS mutations may have prognostic value in pancreatic cancer (Kim et al., 2011; Boeck et al., 2013a). P53 is a transcription factor that is activated in response to apoptosis, cell cycle, and DNA repair. Genetic deletions or mutations of the P53 tumor suppressor gene are prevalent in pancreatic carcinomas (40% to 87% of cases) (Cicenas et al., 2017). Recent evidence showed that mutated P53 causes genetic instability, drives metastasis, and overcomes growth arrest/senescence in pancreatic cancer (Du et al., 2017).
Furthermore, some studies identified that mutated P53 associated with poor prognosis in pancreatic cancer, but inconsistent results were reached (Cicenas et al., 2017). CDKN2A/P16 is a recognized tumor suppressor because of its role in preventing the progression through the G1 cell cycle checkpoint (Hussussian et al., 1994). Shreds of evidence showed that CDKN2A/P16 alterations might participate in the aggressiveness of pancreatic carcinoma (Vasen et al., 2000; Cicenas et al., 2017; Smigiel et al., 2018).
Recent large-scale whole-genome and whole-exome sequencing studies have shown that activating mutations in KRAS are nearly ubiquitous and inactivation of TP53, SMAD4, and CDKN2A/P16 occurs at rates of 50% (Jeong et al., 2005; Burki, 2015; Waddell et al., 2015; Barrett et al., 2017). This finding raised the question whether gene aberrations in cancer cells are important determinants of disease outcome. Moreover, the prognostic value of these driver mutations in pancreatic cancer has not been well defined. The purpose of the current meta-analysis was therefore to clarify the clinical implications of these driver genes in the prognosis of pancreatic cancer.
Materials and Methods
Search strategy
An extensive search of articles and abstracts was conducted in all major electronic databases from inception to June 2018, using the following MeSH terms and text words: (“pancreas” OR “pancreatic”) AND (“markers”) AND (“immunohistochemistry” OR “PCR”) AND (“survival” OR “prognosis” OR “prognostic”). Two reviewers (Yu and Hong) independently conducted a search of EMBASE, MEDLINE, Cochrane Central Register of Controlled Trials, and Cochrane Database of Systematic Reviews, using PubMed and Ovid as search engines, as well as Google Scholar. Each search was repeated for individual markers by substituting the names of markers of interest along with relevant synonyms: “P53” OR “TP53”; “CDKN2A/P16” OR “CDKN2A/P16*” OR “CDKN2A”; “SMAD4” OR “smad-4”; “DPC4” OR “DPC-4”; “KRAS.” Finally, we scrutinized the reference lists of all relevant articles to identify studies that may not have been identified by the strategy outlined above.
Inclusion criteria and exclusion criteria
Articles which passed the primary screening were further scrutinized for the presence of all the following items: (1) Cohort studies or case-control studies, which were concerning the relationship between driver mutations and cancer; (2) Cancer diagnosis must be clearly proved; (3) The hazard ratio (HR) and 95% confident intervals (CIs) were best characterized by sufficient information; and (4) Ruling out such studies which were concerning animals or cell lines and even case-series, letters, editorials, comments, reviews, and abstracts. Those studies that lead us to no detailed information to estimate the HR and 95% CIs were also excluded, even though contacts with relevant authors had been made.
Data extraction
Eligible studies and data were extracted using a standardized data collection form to increase the uniformity of data extraction. The reviewers had a discussion to resolve the inconsistencies between their data until the consistency was reached.
Assessment of study quality
The Newcastle-Ottawa Scale (NOS) was used to assess the quality of included studies. A star system has been developed for the assessment. It is feasible for us to set a study awarded 0 to 3, 4 to 6, or 7 to 9 stars as a low-, moderate-, or high-quality study divided as previously described (Yu et al., 2017).
The selected studies were evaluated independently by two investigators (Yu and Hong) in a blinded manner. Besides, any inconsistency would be solved by a discussion with the third investigators (Gu).
Statistical analysis
We performed meta-analysis using Review Manager (version 5.3 for Windows) and Stata (version 12 for Windows). HR and 95% CI were used to comparing the different impacts of driver status on the survival of patients, whereas OR with 95% CI was applied to assess the association between driver mutations and clinicopathological features. HRs and corresponding 95% CIs of low expression versus high expression were adopted in some studies; then we calculated reciprocal to get high expression versus low expression data (Tierney et al., 2007). In cases of no direct HR and corresponding 95% CI founded in some studies, we used the methods developed by Parmar (Parmar et al. 1998) 45, Williamson (Williamson et al., 2002) 46, and Tierney (Tierney et al., 2007) to estimate these values based on available data, such as survival curves. In our meta-analysis, heterogeneity was assessed by the chi-square test and p-value. The I2 value was used to assess heterogeneity, and if I2 = 0-50%, a fixed-effect model is used, meaning that there is no significant heterogeneity. Otherwise, the random-effect model was applied. The illustration of the HRs and 95% CIs of each included study was shown by forest plots, and the results were pooled. Funnel plots were used to assess evidence for publication bias. Sensitivity analysis was conducted by extraction of every single study to investigate the stability of the results and the resource of heterogeneity. All p-values were two sided, being statistically significant when the p-value <0.05.
Results
Study characteristics
We searched MEDLINE, EMBASE, Cochrane Database of Systematic Reviews, Cochrane Central Register of Controlled Trials, using PubMed and Ovid as search engines, and Google Scholar from inception to June 2018, to identify potentially relevant published literature and yielded 355 studies of P53, 238 of SMAD4, 201 of CDKN2A/P16, and 303 of KRAS. After careful screening, 17 studies on P53 (Ahrendt et al., 2000; Nio et al., 2001; Yamasawa et al., 2002; Hashimoto et al., 2005; Dong et al., 2007; Grochola et al., 2011; Oshima et al., 2013; Shin et al., 2013; Tsukamoto et al., 2013; Kim et al., 2014; Striefler et al., 2016; Wang et al., 2016; Zhao et al., 2016), 8 on SMAD4 (Tascilar et al., 2001; Biankin et al., 2002; Hua et al., 2003; Toga et al., 2004; Ottenhof et al., 2012; Oshima et al., 2013; Shin et al., 2013; Wang et al., 2016), 7 on CDKN2A/P16 (Naka et al., 1998; Kawesha et al., 2000; Gerdes et al., 2002; Ohtsubo et al., 2003; Oshima et al., 2013; Georgiadou et al., 2014; Wang et al., 2016), and 2 on KRAS (Boeck et al., 2013b; Shin et al., 2013) met our eligibility criteria (Fig. 1). The detailed information of included studies is summarized in Table 1.
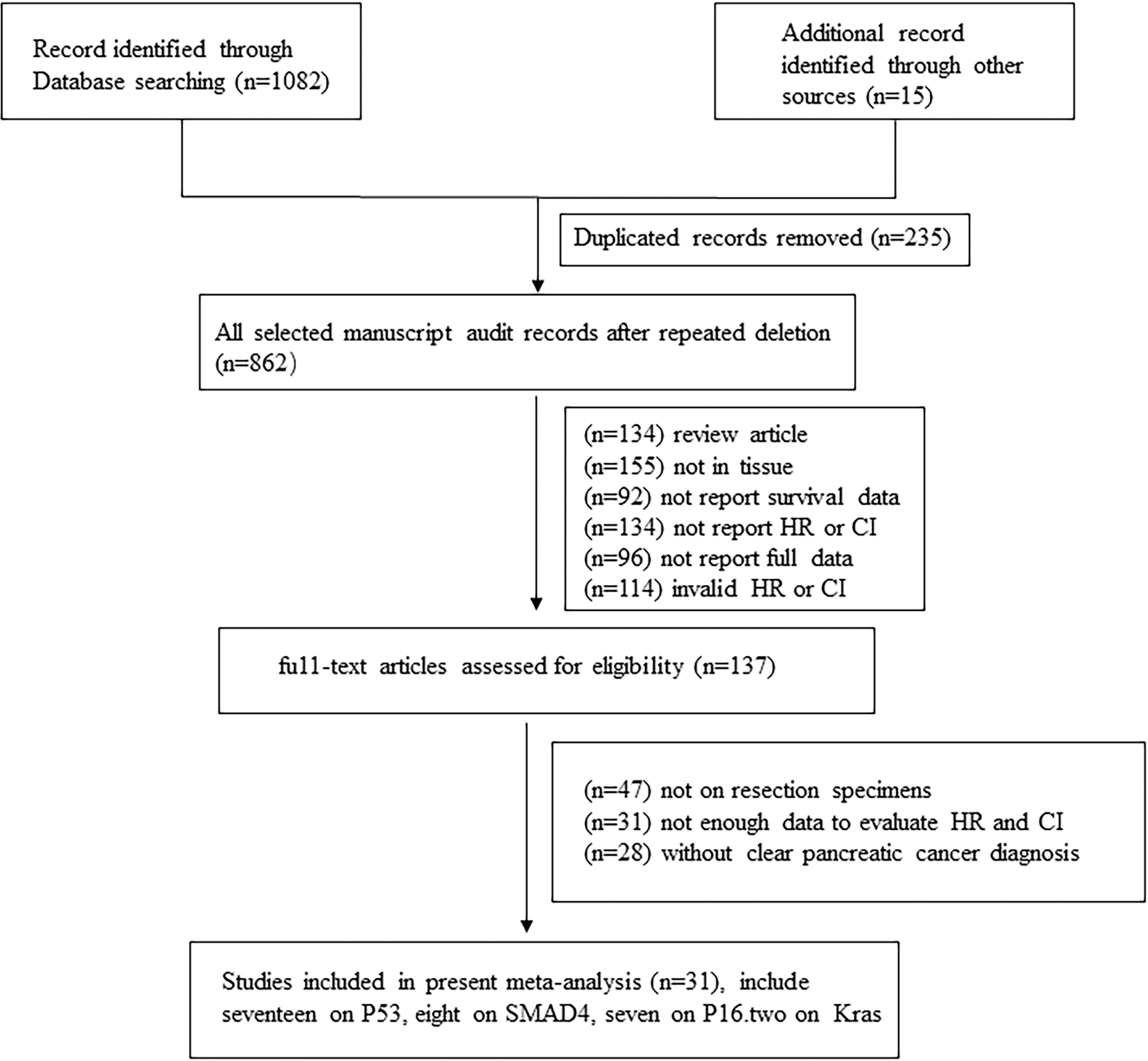
Flow diagram of study selection.
Basic Information of Included Studies
DFS, disease-free survival; PFS, progression-free survival; RFS, relapse free survival; N.S., not significant; OS, overall survival.
Briefly, all eligible studies were retrospective studies that contained 3373 samples and published from 1999 to 2018. All the included studies measured the expression of gene aberrations utilizing immunohistochemistry (IHC) staining, but the cutoff values varied across studies.
Seventeen studies provided information on the association between P53 level and overall survival (OS) (Bold et al., 1999; Ahrendt et al., 2000; Nio et al., 2001; Yamasawa et al., 2002; Hashimoto et al., 2005; Dong et al., 2007; Grochola et al., 2011; Oshima et al., 2013; Shin et al., 2013; Tsukamoto et al., 2013; Kim et al., 2014; Ormanns et al., 2014; Sheng et al., 2014; Wang et al., 2015, 2016; Striefler et al., 2016; Zhao et al., 2016), 8 on association between SMAD4 level and OS (Tascilar et al., 2001; Biankin et al., 2002; Hua et al., 2003; Toga et al., 2004; Ottenhof et al., 2012; Oshima et al., 2013; Shin et al., 2013; Wang et al., 2016), 7 on association between CDKN2A/P16 level and OS (Naka et al., 1998; Kawesha et al., 2000; Gerdes et al., 2002; Ohtsubo et al., 2003; Oshima et al., 2013; Georgiadou et al., 2014; Wang et al., 2016), and 2 on association between KRAS level and OS (Boeck et al., 2013b; Shin et al., 2013). Studies by Striefler (Striefler et al., 2016) reported disease-free survival data, whereas the study by Wang (Wang et al., 2016) reported data on relapse-free survival. Only one study reported data on progression-free survival (Ormanns et al., 2014).
Although we had used different keywords and got 303 studies from PubMed, EMBASE, and ISI Web of Science, there were only 2 eligible studies (Boeck et al., 2013b) on KRAS searched. We found that there was great significance to evaluate the prognostic value of KRAS in pancreatic cancer. KRAS gene is a recognized oncogene, which affects the EGFR-mediated cell pathway. Ras gene products are involved in the kinase signaling pathways that control gene transcription, which regulates cell growth and differentiation, resulting in overexpression and amplification of Ras, and this is a crucial step in tumorigenesis (Hashimoto et al., 2005; Boone et al., 2014). Therefore, we still concluded and discussed the expression of KRAS in pancreatic cancer.
The association between clinicopathological variables and survival reported in the included studies is summarized in Table 1. Among the studies, 9 studies on p53 (Ahrendt et al., 2000; Yamasawa et al., 2002; Hashimoto et al., 2005; Dong et al., 2007; Grochola et al., 2011; Tsukamoto et al., 2013; Kim et al., 2014; Wang et al., 2015; Zhao et al., 2016), 4 on SMAD4 (Tascilar et al., 2001; Biankin et al., 2002; Hua et al., 2003; Toga et al., 2004), and 4 on CDKN2A/P16 (Ohtsubo et al., 2003; Wang et al., 2016) evaluated the tumor differentiation.
Overall analyses
In this review, 3373 patients of 31 studies in pancreatic cancer were included to estimate the prognostic influence of overexpression of P53, loss of SMAD4 and CDKN2A/P16, and KRAS mutation. Statistics demonstrated that 2119 patients with overexpression of TP53 mutation had shorter overall survival with a HR of 1.249 (95% CI = 1.003-1.554, p = 0.047). There was median-moderate heterogeneity across studies with significant statistics (I2 = 60.4%, p = 0.001) (Ahrendt et al., 2000; Nio et al., 2001; Yamasawa et al., 2002; Hashimoto et al., 2005; Jeong et al., 2005; Dong et al., 2007; Grochola et al., 2011; Oshima et al., 2013; Shin et al., 2013; Tsukamoto et al., 2013; Kim et al., 2014; Wang et al., 2015; Wang et al., 2016; Zhao et al., 2016) (Fig. 2A), and the random-effect model was used. The abnormal expression of CDKN2A/P16 showed no significant association with survival with the HR of 0.916 (95% CI = 0.583-1.439, p = 0.704). The heterogeneity was across studies (I2 = 75.2%, p heterogeneity <0.001) (Fig. 2B).
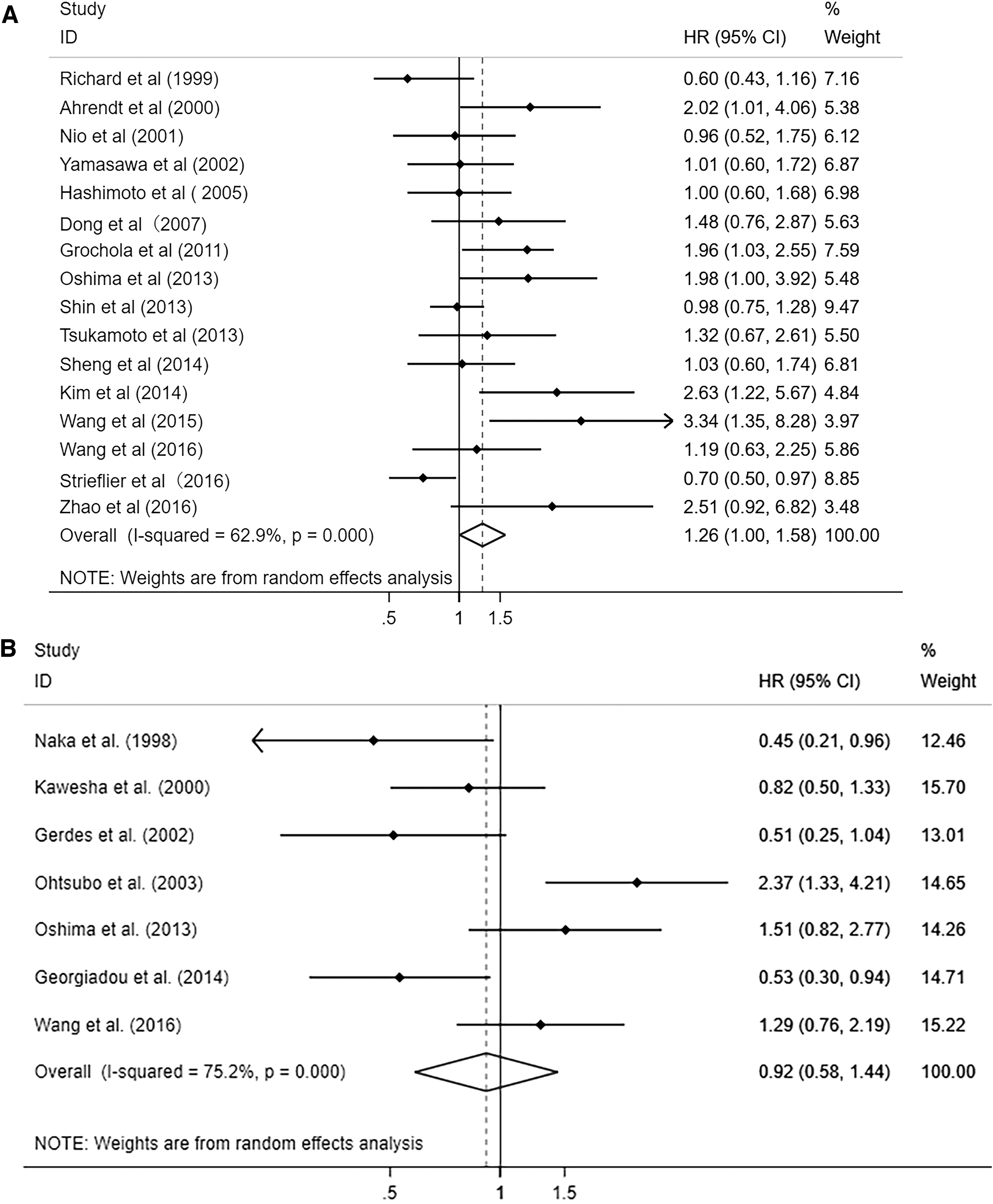
Forest plot of HR for the association between OS and P53
The loss of SMAD4 expression had a statistically significant adverse effect (Tascilar et al., 2001; Biankin et al., 2002; Hua et al., 2003; Toga et al., 2004; Ottenhof et al., 2012; Oshima et al., 2013; Shin et al., 2013; Wang et al., 2016) on overall survival with a HR of 1.397 (95% CI = 1.015-1.922, p = 0.040) (Fig. 3A). Heterogeneity of SMAD4 was statistically significant (I2 = 62.6%, p heterogeneity = 0.009). The random-effect model was applied. Although there were only two included studies on KRAS mutation, we still presented quantitative analysis and results demonstrated that the pancreas patients of KRAS mutation had negative prognostic influence with the HR of 1.68 (95% CI = 1.27-2.22, p < 0.001). The heterogeneity was across studies (I2 = 0.0%, p heterogeneity = 0.992) (Fig. 3B). Shin and collaborators (Tascilar et al., 2001; Biankin et al., 2002; Hua et al., 2003; Toga et al., 2004; Ottenhof et al., 2012; Oshima et al., 2013; Shin et al., 2013; Wang et al., 2016) presented that KRAS mutational status was an independent factor influencing the prognosis of pancreas.
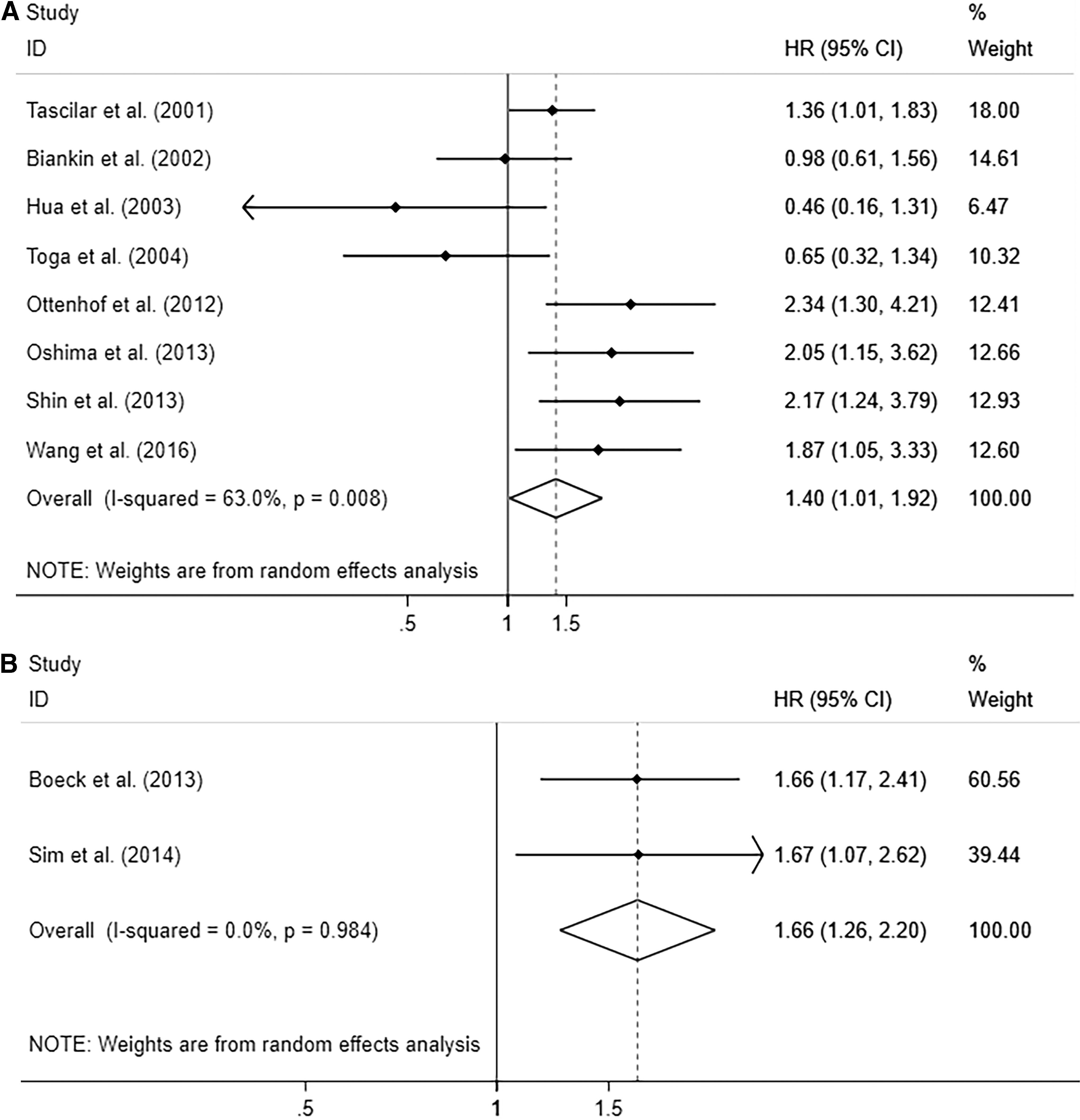
Forest plot of HR for the association between OS and SMAD4
Correlation of driver mutations with clinicopathological features
Overexpression of p53 had significant association with vascular invasion (OR = 1.97, 95% CI = 1.14-3.40; I2 = 0.0%, p = 0.696) (Supplementary Fig. S1A). Overexpression of p53 appeared to be not significantly associated with age (OR = 1.12, 95% CI = 0.62-2.02; I2 = 66.3%, p = 0.007) (Supplementary Fig. S1B), gender (OR = 0.89, 95% CI = 0.68-1.18; I2 = 0.0%, p = 0.845) (Supplementary Fig. S1C), positive lymph metastasis (OR = 1.36, 95% CI = 0.90-2.11; I2 = 42.1%, p = 0.097) (Supplementary Fig. S1D), and tumor stage (UICC) (OR = 0.78, 95% CI = 0.46-1.32; I2 = 48.0%, p = 0.052) (Supplementary Fig. S1E). As for SMAD4, its abnormal expression did not correlate with gender (OR = 0.97, 95% CI = 0.62-1.52; I2 = 0.0%, p = 0.753) (Supplementary Fig. S1F) and positive lymph metastasis (OR = 0.79, 95% CI = 0.24-2.65; I2 = 69.8%, p = 0.036) (Supplementary Fig. S1G). And loss of CDKN2A/P16 was identified to not be associated with positive lymph metastasis (OR = 1.97, 95% CI = 1.14-3.40; I2 = 0.0%, p = 0.696) (Supplementary Fig. S1H).
Heterogeneity
We conducted subgroup analyses and sensitivity analyses to explore the sources of the heterogeneity. According to ethnicity, sample size, recruitment time, antibody source, study quality, and the analyses of univariate or multivariate, the included studies of p53, CDKN2A/P16, and SMAD4 were divided into two subgroups. Because there are only two studies of KRAS, it is not necessary to explore the heterogeneity. However, these variables did not modify the prognostic value of p53, p16, and SMAD4.
The subgroup results are shown in Table 2: A significant relationship between P16 expression and OS in subgroup from non-Asian (HR = 0.641, 95% CI = 0.461-0.891, p = 0.408; I2 = 0%) (Supplementary Fig. S2). For P53, it was significantly associated with the overall survival in the subgroup of low study quality (HR = 1.350, 95% CI = 1.023-1.782, p = 0.003; I2 = 61.6%), late recruitment time (HR = 1.396, 95% CI = 1.037-1.879, p = 0.001; I2 = 66.8%), and the subgroup of multivariate analysis (HR = 1.329, 95% CI = 1.041-1.697, p = 0.003; I2 = 58.7%) (Supplementary Fig. S3). The association between SMAD4 expression and OS was also observed among the subgroup of high study quality (HR = 2.184, 95% CI = 0.1.449-3.29, p = 0.747; I2 = 0%) and the subgroup of another antibody source (HR = 2.115, 95% CI = 1.518-2.946, p = 0.863; I2 = 0%) (Supplementary Fig. S4).
Results of Subgroup Analysis of Pooled Hazard Ratios of Overall Survival of Patients with Abnormal Driver-Mutation Level
CI, confident interval.
Sensitivity analyses
To estimate the stability of the studies, we conducted sensitivity analyses by assessing the potential impact of the individual study on the pooled data of every gene. The pooled results of the studies of p16, p53, KRAS, and SMAD4 were not affected after excluding every single study of these genes separately, which indicated the stability of the present results (Supplementary Fig. S5).
Publication bias
Begg's test, Egger's test, and funnel plot were applied to estimate the publication bias of all these studies. The shape of the funnel plot and the results of Begg's test (CDKN2A/P16, p = 0.764; p53, p = 0.308; SMAD4, p = 0.027) showed no publication bias in all included genes (Supplementary Figs. S6A-C and S7A-C). Publication bias was found in the results of Egger's test in the study of CDKN2A/P16, p53, and SMAD4 (CDKN2A/P16, p = 0.393; p53, p = 0.151; SMAD4, p = 0.021) (Supplementary Fig. S7E-G). Considering the non-normal distribution of the included patient numbers and the deviation between Begg's test and Egger's test, the results of the Egger's test were not trusted. Because there are only two studies of KRAS, the publication bias was not considered. For prognosis, we still conducted the funnel plot, Begg's test, and Egger's test, which indicated no publication bias on KRAS (Supplementary Figs. 6D, 7D, 7H). Consequently, there was no publication bias in all the above studies. In addition, it presented to us that the genes of p16, p53, SMAD4, and KRAS had essential influences in the prognosis of pancreatic cancer.
We used the corresponding survival data from TCGA dataset (Peran et al., 2018; Nicolle et al., 2019) to analyze (Peran et al., 2018; Nicolle et al., 2019) and explore the association between TP53, P16, SMAD4, and KRAS expression and PADC patients' clinical outcome (Supplementary Fig. S8). An online website TIMER is used for analysis (http://timer.cistrome.org/). This database is based on TCGA database, and this website is also recognized as a relatively authoritative website at present (Li et al., 2017, 2020). The results showed that SMAD4 and KRAS expression correlates strongly with the OS [Supplementary Fig. S8: p (SMAD4) = 0.036, p (KRAS) = 0.007]. Moreover, it was found that TP53 and P16 have no obvious effects on the prognosis of the disease [Supplementary Fig. S8: p (P16) = 0.847, p (TP53) = 0.934]. However, in our study, we found that these four genes are associated closely with the prognosis of pancreatic cancer. The differences between the two results may be due to different experimental methods used in these included articles. In other words, all the articles included in this article adopted IHC or real-time quantitative PCR detecting system. The data from the TCGA database are based on the results obtained from sequencing. Above all, we haven't found the significance of these four genes in clinical treatment. In short, PDAC has an extremely complex disease developmental process, which involves the multimolecular regulation mechanism. We believe that more driver genes should be explored to the best of our ability, not limited to KRAS, CDKN2A/P16, TP53, and SMAD4.
Discussion
Pancreatic cancer continues to be one of the most challenging malignancies despite vigorous research endeavors (Grenacher and Juchems, 2017). Consequently, there is an urgent need to better select patients for current therapies and develop novel therapeutic strategies.
Pancreatic cancer progression is thought to be driven by accumulating genetic alterations (Oshima et al., 2013) (including those in K-Ras, TP53, SMAD4, and CDKN2A/P16) and subsequent signal pathway change, leading to increased cancer cell proliferative potential (Oshima et al., 2013). However, gene aberrations have not yet been used to predict prognosis in pancreatic cancer independently. Herein, we discuss the prognostic value of driver mutations in pancreatic adenocarcinoma and related therapeutic opportunities.
In the present study, 17 studies on P53, including 2200 patients (Ahrendt et al., 2000; Nio et al., 2001; Yamasawa et al., 2002; Hashimoto et al., 2005; Jeong et al., 2005; Dong et al., 2007; Grochola et al., 2011; Oshima et al., 2013; Shin et al., 2013; Tsukamoto et al., 2013; Kim et al., 2014; Wang et al., 2015; Wang et al., 2016; Zhao et al., 2016), 8 studies on SMAD4, including 1162 patients (Tascilar et al., 2001; Biankin et al., 2002; Hua et al., 2003; Toga et al., 2004; Ottenhof et al., 2012; Oshima et al., 2013; Shin et al., 2013; Wang et al., 2016), and 7 studies on CDKN2A/P16 involving 739 patients (Naka et al., 1998; Kawesha et al., 2000; Gerdes et al., 2002; Ohtsubo et al., 2003; Oshima et al., 2013; Georgiadou et al., 2014; Wang et al., 2016) were extracted to evaluate the association between these gene aberrations and prognosis of pancreatic cancer patients. The poor results had demonstrated that the overexpression of P53, loss of SMAD4 and CDKN2A/P16, and KRAS mutation in the resected biological specimen from pancreatic cancer patients predicted the poorer overall survival. We conducted a sensitivity analysis and found that the combined HR did not change significantly.
In the study of Biankin and his colleague (Biankin et al., 2002), preoperative assessment of SMAD4 expression has potential as a prognostic indicator in patients with pancreatic cancer (Biankin et al., 2002). Results showed that resection did not benefit those patients whose cancer expressed SMAD4, and preoperative assessment of SMAD4 expression was beneficial for adequate treatment strategy. Accurate assessment of SMAD4 expression by tumor biopsy, unlike tumor size, margin status, and the peripheral invasion, does not require resection. In addition, in the study by Ohtsubo and others, there was no significant correlation between the expression of CDKN2A/P16 protein and any of the clinicopathological parameters. However, there was a tendency for the tumor to be larger in patients with decreased expression of CDKN2A/P16 protein than those with normal levels.
In contrast, the tumor was significantly larger, and the survival period was significantly shorter for patients with CDKN2A/P16 mutation or hypermethylation than for those with an intact CDKN2A/P16 gene (p > 0.05). The normal or even overexpression of SMAD4 in pancreatic cancer patients usually predicts a better prognosis than those with decreased expression or loss of the gene. More clinical trials for the preoperative assessment of SMAD4 in pancreatic cancer should be conducted, and thus, it may lead the doctors to select better and all-round projects of cure. Besides, the difference between the results of meta-analysis and TCGA may be caused by the different sample numbers included in them. In the future, more research methods may be needed to study and to explore whether CDKN2A/P16 alterations participate in the aggressiveness of pancreatic carcinoma and be used to predict the growth of the tumor and direct the clinicians to assess the character of the tumor better and select adequate adjuvant therapy.
More trials of the assessment of P53 should be proposed, and thus, it may be helpful to identify patients with highly metastatic pancreatic cancer who may benefit from aggressive therapeutic intervention. Sadly, FDA approved therapies that exploit these genomic alterations in pancreatic cancer are currently not available (Liang et al., 2012; Sandhu et al., 2016). Nowadays, there are many different technologies that are changing clinical practice, including the cDNA microarray, in situ fluorescence hybridization, and quantitative reverse transcriptase-polymerase chain reaction. However, these technologies are still in the laboratory. During the process of data collection in the present analysis, we found that IHC was adopted by many people and was simpler and more feasible than other methods. It is easy to use EUS-FNA specimens to assess the molecular expression status of patients with PDAC before surgery. Pancreatic cancer is a disease at the genetic level. Stratifying patients at the genetic level is valuable for future clinical trials. Besides, it can provide enough power to help clinicians diagnose patients with PDAC. For example, if a patient has multiple mutations, the value of performing an extended resection may be doubtful. As results found in our analysis showed that patients with mutations of driver genes had poor survival, patients may not benefit from surgery. The study from Oshima et al. indicated that the number of gene mutations is closely related to the prognosis. A variety of genes can be used to predict the prognosis of patients. Counting the gene mutations may be valuable in prognosis prediction. However, there are few articles and insufficient data. So, there is no way to conduct a meta-analysis of the prognostic value of mutation number, which may be the research direction in the future.
At present, we have found that these four genes are associated with the prognosis of pancreatic cancer. However, we are yet to find its significance in clinical treatment. PDAC has an extremely complex disease development process, which involves the multimolecular regulation mechanism. Therefore, we believe that more driver genes should be explored as much as possible, not limited to KRAS, CDKN2A/P16, TP53, and SMAD4. We counted almost 180 cases based on the TCGA database. It was found that only TP53 and SMAD4 affected the prognosis of the disease. We suggest that the conflicting results may be due to different experimental methods, either using RNA sequencing or IHC. Therefore, we believe that the prognosis of pancreatic cancer patients should be determined not only by the four genes mentioned in this article but also by other genes.
Nevertheless, we acknowledge that this study has some limitations. First, the methods of detecting the expression of the markers are not the same. Two studies of KRAS, 5 studies of CDKN2A/P16, 8 studies of SMAD4, and 13 studies of P53 were reported to use IHC, of which the dilution and primary antibody were different. Second, the definition for positive P53, CDKN2A/P16, SMAD4, and KRAS was different; for instance, some studies preferred the percentage of positive cells, and others chose both staining intensity and the percentage of positive cells. Third, the approach of HR extraction may result in a potential influence. In all the studies evaluating the relationship between markers' expression and overall survival, HRs and 95% CIs were reported directly in some studies, and HRs and 95% CIs were extracted from the Kaplan-Meier survival curves in other studies. Nevertheless, the method of extrapolating HR from survival curves may be less accurate than the direct method.
The meta-analysis has previously proved to overexpress these four markers, P53, CDKN2A/P16, SMAD4, and KRAS, to predict poor overall survival in patients with pancreatic cancer. However, their research is limited. For example, in 2011 Smith made a similar meta-analysis, but his data have certain limitations due to time. We have also considered a novelty when discussing whether we need to carry out this study. However, we believe that with the advancement of medical technology and the increase of research data, it is timely to have a new meta-analysis to calculate the current data analyses. We put forward a highlight of this article, which is exploited by multiple genes to analyze the prognosis of patients with pancreatic cancer comprehensively. Current evidence showed that assessment of combinations of genetic alterations is presumably more predictive of survival outcome compared with single changes. However, few studies concerning this issue were performed, and no pooled results were available.
Conclusions
The pooled results have demonstrated that the overexpression of P53, loss of SMAD4, and KRAS mutation in the resected biological specimen from pancreatic cancer patients predicted the poorer overall survival, and this effect is independent of tumor stage and other factors.
Footnotes
Ethical Approval
This article does not contain any studies with human participants performed by any of the authors.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This article is supported by Medical Scientific Research Foundation of Guangdong Province, China under (Grant No. B2020225) and Supported by Science and Technology innovation of Zhaoqing City, Guangdong Province, China under (Grant No. 2019N008).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.