
Abstract
Objective:
Osteogenesis imperfecta (OI) includes a group of disorders characterized by susceptibility to bone fractures with different severities. The increasing number of genes that may underlie the disorder, along with the broad phenotypic spectrum that overlaps with other skeletal diseases, provided a compelling case for the use of high-throughput sequencing (HTS) technology as an aid to OI diagnoses. The aim of this analysis was to present the data from our 5-year targeted HTS results, that includes the reporting of 9 novel and 24 known mutations, found in OI patients, from 5 different regions of Turkey.
Materials and Methods:
We performed a retrospective cross-sectional study, reporting the HTS results of 43 patients (23 female and 20 male; mean age: 9.5 years), directed to our center with a suspicion of OI between February 2015 and May 2020. Genetic analyses were also performed for 24 asymptomatic parents to aid the segregation analyses. We utilized an HTS panel targeting the coding regions of 57 genes associated with a reduction, increase, or abnormal development of bone mineralization. In addition, we sequenced the entire coding region of the IFITM5 gene through HTS.
Results:
Thirty-nine patients had at least one pathogenic/likely pathogenic variation (90.69%) in the COL1A1 (56.41%), COL1A2 (20.51%), FKBP10 (7.7%), P3H1 (5.13%), IFITM5 (5.13%), CTRAP (2.56%), or TMEM38B (2.56%) genes. Nine of the determined pathogenic/likely pathogenic variations were novel. The recurrent pathogenic mutations were c.1081C>T (p.Arg361Ter) (3/43), c.1405C>T (p.Arg469Ter) (2/43), and c.3749del (p.Gly1250AlafsTer81) in COL1A1 gene, along with c.-14C>T variation in the 5’UTR of the IFITM5 gene (2/43) and the c.890_897dup variation in the FKBP10 gene (2/43). Three out of 43 patients were carrying at least one additional variant of unknown significance, highlighting the importance of a multigene panel approach and segregation analyses.
Conclusion:
We suggest that a targeted HTS panel is a feasible tool for genetic diagnosis of OI in patients.
Introduction
Osteogenesis imperfecta (OI), also known as brittle bone disease, is a group of clinically and genetically heterogeneous disorders, characterized by susceptibility to bone fractures with different severity (Marini et al., 2017). There is a large spectrum of other clinical manifestations, in addition to bone fragility, including short stature, dentinogenesis imperfecta, blue sclera, conductive or sensorineural hearing loss, scoliosis, pulmonary function impairment, cardiac valve abnormalities, muscle weakness, and ligamentous laxity (van Dijk et al., 2011; Marini et al., 2017). The incidence of OI is about 1/15-20,000 (Valadares et al., 2014).
The clinical spectrum of OI ranges from severe types, causing perinatal lethal form, to the milder phenotypes, which can manifest in adulthood. The clinical classification of OI was first formulated by Sillence in 1979 (Sillence et al., 1979). In this first classification system, the disease was divided into four types: OI type I was the mild OI with blue sclerae, type II was the lethal perinatal OI, type III was the progressively deforming OI with normal sclerae and type IV was the OI with dentinogenesis imperfecta (Valadares et al., 2014). The first two genes implicated in the pathogenesis of OI were COL1A1 (+ 120150) and COL1A2 (* 120160) genes, which are responsible for the majority of dominantly inherited OI (Pope et al., 1985). Further studies revealed that there are many more different types of the disease (Valadares et al., 2014).
In the last decade, defects across a wide range of genes encoding proteins that are involved in the construction of bone and differentiation of bone-forming cells have been determined as the underlying cause of the different types of OI (Marini et al., 2017). The inheritance can be both dominant and recessive, depending on the genes responsible for the disease (Valadares et al., 2014). Although a new classification, including genetic basis, was created due to the growing list of OI-related genes, there are still some debates on the use of this novel expanded classification system (Basel and Steiner, 2009; van Dijk et al., 2011; Valadares et al., 2014; Marini et al., 2017; Tauer et al., 2019).
The genetic diagnosis of OI patients is challenging because of the phenotypic spectrum that may overlap with other skeletal diseases (Trancozo et al., 2019). High-throughput sequencing (HTS) facilitates the simultaneous analysis of multiple genes independent from their size, and thus is superior to conventional methods like Sanger Sequencing in terms of speed and cost-effectiveness of the diagnosis of genetically heterogeneous disorders like OI (Sule et al., 2013; Le Gallo et al., 2017). However, classification of the rare variants found as a result of HTS is challenging (Garrett et al., 2016; Vears et al., 2017). Functional studies are time-consuming and are not practical in clinical situations, where there is a shorter turnaround time for reporting the results. In silico prediction tools, guidelines, databases, and study results together are being used for interpreting the results (Tang et al., 2019). Standards and Guidelines like ACMG 2015 are very helpful for global consensus on the determination of disease-related pathogenic/likely pathogenic variations (Richards et al., 2015).
The accumulation of evidence on the pathogenic variants across different patient populations provides important guidance for human geneticists (Duzkale et al., 2013; Wang et al., 2015). In this study, we present our results of targeted HTS, including 9 novel pathogenic/likely pathogenic variations found in the brittle bone-related genes of 43 OI patients from 5 different regions of Turkey. To our knowledge, besides the novel variations found, this is the first comprehensive report of high-throughput sequencing results of OI patients from Turkey.
Materials and Methods
Samples
This retrospective cross-sectional study has been approved by the Research Ethics Board of Trakya University's Faculty of Medicine (TUTF-BAET 307/2020). Targeted HTS results of 67 individuals analyzed for OI-related genes between February 2015 and May 2020 are included in this study. Forty-three of those individuals were index cases (23 female and 20 male; mean age: 9.95 ± 12.02 years) directed to Trakya University Hospital, Department of Medical Genetics, Genetic Diseases Diagnosis Center, with the clinical suspicion of OI. The remaining 24 individuals were available asymptomatic parents tested for segregation analysis. Index patients were from 5 different cities of Tukey (Edirne [21 samples], Bursa [2 samples], Diyarbakır [13 samples], Samsun [6 samples], and Kayseri [1 sample]). Patient demographic and clinical features are summarized in Table 1.
Summary of Patient Demographic and Clinical Features
OI, osteogenesis imperfect.
We collected EDTA blood samples, after the written informed consent forms were taken from the patients or their legal guardians. Genomic DNA was isolated from peripheral white blood cells of the patients by using the EZ1 DNA Investigator Kit (Qiagen, Hilden, Germany). We performed primary quality control of the isolated DNA samples using NanoDrop (Thermo Fisher Scientific, Waltham, MA). Around 260/280 ratios of DNA samples were between 1.8 and 2.0 and 260/230 ratios were between 2.0 and 2.2. Qubit fluorometer was used as the second quality control step before HTS.
Library preparation for targeted multigene HTS
Osteo-GeneSGKit DensidadOsea, IVD -CE, including 57 genes (ALPL, FGFR1, CYP27B1, ANKH, RASGRP2, DLX3, LEPRE1[P3H1], CA2, LEMD3, SLC34A1, BANF1, COL1A1, CTSK, EXT1, TNFSF11, SQSTM1, TCIRG1, SLC9A3R1, GORAB, TNFRSF11B, PPIB, TREM2, LRP5, TNFRSF11A, CRTAP, TMEM38B, CLCN7, OSTM1, SERPINH1, TYROBP, PTH1R, SLC34A3, MMP2, GJA1, WNK1, TGFB1, PLOD2, ANO5, SERPINF1, ENPP1, FGF23, GNAS, SH3BP2, EXT2, FKBP10, COL1A2, WNT1, PHEX, DMP1, LRP4, SOST, BMP1, SP7, AMER1, HPGD, FERMT3, and PLEKHM1) (Fig. 1), targeting diseases associated with a reduction, increase, or abnormal development of bone mineralization was used for analysis of 23 individuals tested between 2015 and 2018. We used QIAseq Targeted DNA Panel (Qiagen), containing the same gene set with the previous kit, for the analysis of individuals tested between 2018 and 2020. QIAseq Targeted DNA Panel (Qiagen) was preferred afterward because it was more cost-effective. Both library kits were designed to cover the entire coding regions of target genes with a 20 bp intron padding.
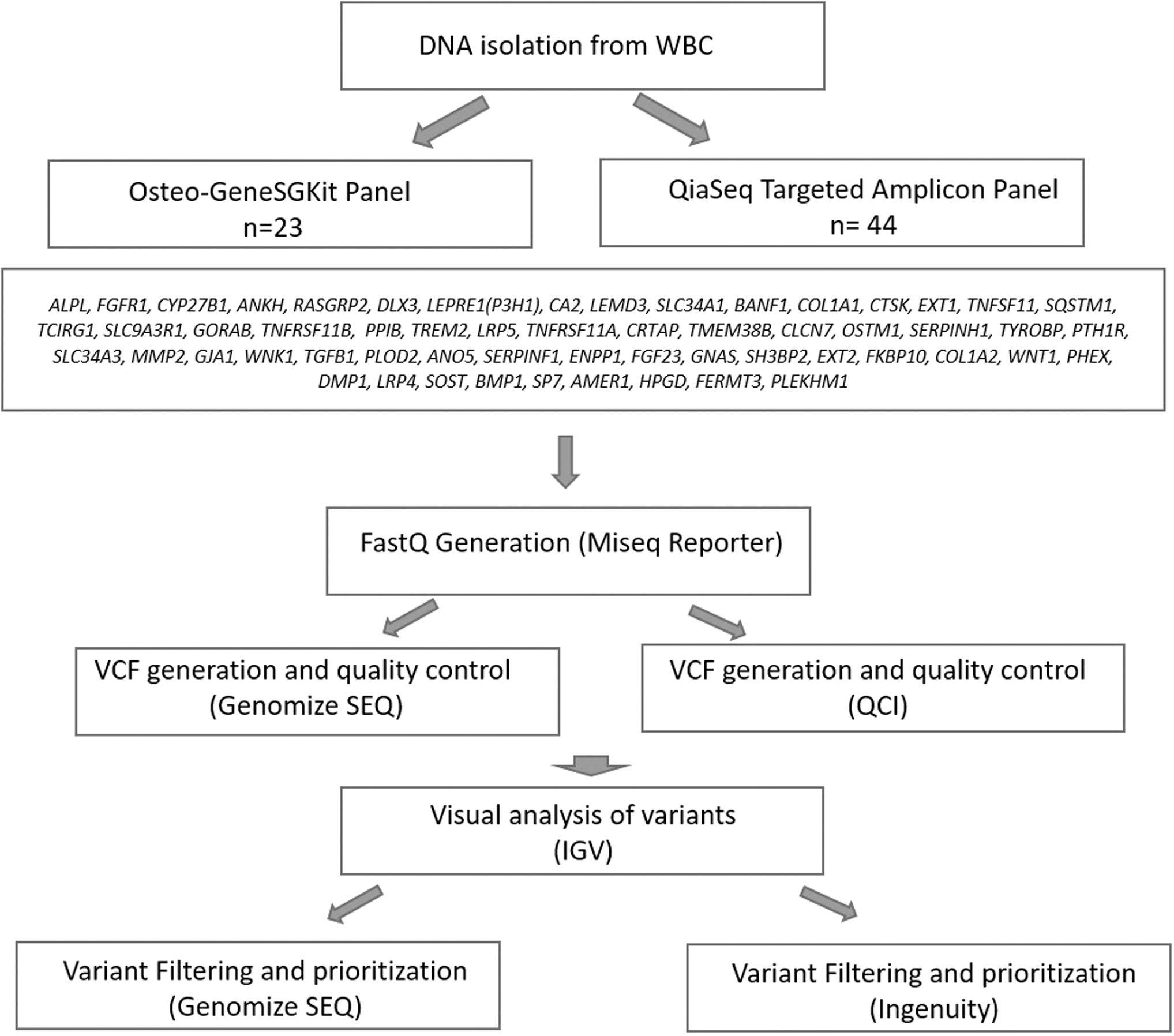
Summary of the targeted HTS workflow applied to our patient population. HTS, high-throughput sequencing.
Sequencing and Fastq generation were performed on MiSeq Reporter (v2.5.1; Illumina, Inc.) for all samples. Genomize Seq (Genomize, İstanbul, Turkey) and QCI (Illumina, San Diego, CA) platforms were used for quality control, vcf generation, and variant filtering/analysis of samples (Fig. 1). At least 20X read depth were accepted as reliable for target regions. Library preparation step was repeated for the samples below the quality parameters. All variants were visually controlled using IGV 2.4.8 (Robinson et al., 2017).
Sequencing of the IFITM5 gene
IFITM5 gene was not included in the targeted amplicon panel kits, so we designed primers to amplify the coding region of the IFITM5 gene in 43 patients, along with two asymptomatic parents—for segregation analysis—by long-range Polymerase Chain Reaction, and sequenced the entire gene using Nextera XT (Illumina) sequencing kit. For this, the entire coding regions of the IFITM5 gene were amplified using a single set of in-house designed IFITM5 primers (forward primer: 5′-GAC ACC TCC AAC TCT TCT TGG CCT-3′ and reverse primer: 5′-TGT GGC ATT TGG CTT TGG TGG TC-3′). The amplicons were pooled and purified using the Agencourt AMPure XP system (Beckman Coulter, Pasadena, CA). The starting DNA library was quantified using the Qubit dsDNA BR Assay kit (Invitrogen, Carlsbad, CA). Sequencing library construction was performed by Nextera XT kit (Illumina, Inc.), which uses transposon to fragment the ends and simultaneously adds adapters and barcoding sequences. The pooled and barcoded libraries were subsequently sequenced on the MiSeq sequencer (Illumina, Inc.). Moreover, Fastq and vcf generation were performed on MiSeq Reporter (v2.5.1; Illumina, Inc.). Amplicons were visually analyzed using IGV 2.4.8.
Data analysis and variant classification
Variants were first filtered according to their population frequencies in gnomAD (Karczewski et al., 2020), 1000 Genome (Auton et al., 2015), and Exac (Karczewski et al., 2017) databases. Variants below 0.5% were further analyzed for their pathogenicity according to ACMG 2015 guidelines. ClinVar (Landrum et al., 2018), dbSNP (Sherry et al., 2001), and HGMD database (Stenson et al., 2017) access were also considered (Table 2).
Annotations, In Silico Predictions, and Previous Database Access Number Information of Each Variant Found in This Study
NA, not available; VUS, variant of unknown significance.
In silico predictions
CADD (Rentzsch et al., 2019) and Mutation Taster (Schwarz et al., 2014) were used to predict the pathogenic potential of all rare/novel variants. SIFT (Ng and Henikoff, 2003) and Polyphen predictions (Adzhubei et al., 2013 ) were used to compare the damaging potential of missense variants. GERP scores (Davydov et al., 2010) and PhyloP (Pollard et al., 2010) were considered for conservation scores. MaxEntScan (Eng et al., 2004) and Gene Splicer (Pertea et al., 2001) predictions were applied to the splicing variants (Table 2). Final classification of variants was made according to ACMG 2015 guidelines, taking into consideration all available information from databases, literature reports, and segregation analysis whenever possible.
Statistical analysis
Descriptive analyses of mutations and patient demographics were performed according to the frequency distributions. Mean age and standard deviation were calculated with R software.
Results
Thirty-nine out of 43 patients had at least one pathogenic/likely pathogenic variation (90.69%) in the COL1A1 (56.41%), COL1A2 (20.51%), FKBP10 (7.7%), P3H1 (5.13%), IFITM5 (5.13%), CTRAP (2.56%), and TMEM38B (2.56%) genes (Table 3). Seventy-six percent of these variations were in the COL1A1 and the COL1A2 genes (Fig. 2). Three out of 39 patients had at least 1 variation of unknown significance together with a pathogenic/likely pathogenic variation. Results of each patient are summarized in Table 1.

Mutation distribution and corresponding domain structure of COL1A1 and COL1A2 exonic regions. Empty arrows indicate VUS variants. SP, signal peptide; VUS, variant of unknown significance.
OI Types, Segregation Analysis, and Final Interpretation of Pathogenicity of the Variants Found in Patients
Nine of the pathogenic/likely pathogenic variations determined were novel, and seven out of these nine novel variations (77.77%) were located on the COL1A1 and COL1A2 genes.
There were five different recurrent pathogenic/likely pathogenic variations located in three different genes in our patient population as follows: NM_000088.3(COL1A1): c.1081C>T (p.Arg361Ter) variation in 17th exon of COL1A1 gene (7.69%); NM_000088.3(COL1A1):c.1405C>T (p.Arg469Ter) variation in the 21th exon of COL1A1 gene (5.12%), NM_000088.3(COL1A1): c.3749del (p.Gly1250AlafsTer81) variation in 48th exon of the COL1A1 gene (5.12%), NM_001025295.3(IFITM5):c.-14C>T variation in the 5′ untranslated region (5′UTR) of the IFITM5 gene (5.12%), and NM_021939.3(FKBP10):c.890_897dup (p.Gly300Ter) variation in the FKBP10 gene (5.12%).
The patient B32 had a heterozygous likely pathogenic NM_022356.3(P3H1):c.2055 + 18G>A variation. This patient had an additional NM_022356.3(P3H1):c.1667A>G variation and segregation analysis in the family revealed that these two mutations were in trans positions; therefore, the patient was compound heterozygous. However, NM_022356.3(P3H1):c.1667A>G variation was a variant of unknown significance (VUS) according to ACMG Guidelines (Richards et al., 2015).
The patient B12 had two different single missense changes: NM_000089.3 (COL1A2): c.671G>A (p.Arg224His) in COL1A2 and NM_000088.3(COL1A1): c.1040G>A (p.Gly347Asp) in COL1A1. Segregation analysis revealed that the first mutation was a de novo likely pathogenic mutation, while NM_000089.3(COL1A2):c.671G>A (p.Arg224His) was inherited from the unaffected mother. Hence, NM_000088.3(COL1A1): c.1040G>A (p.Gly347Asp) missense likely pathogenic variants in the COL1A1 gene were suggested to be responsible for the disease in this patient.
In the patient B40, there was a novel homozygous likely pathogenic NM_018112.3(TMEM38B):c.112 + 1dupG variation. The patient also had both heterozygous NM_000088.3(COL1A1):c.77G>A (p.Gly26Asp) and heterozygous NM_000089.3(COL1A2):c.3473A>C (p.Asn1158Thr) variations. Segregation analysis revealed that NM_000088.3(COL1A1):c.77G>A (p.Gly26Asp) variation was inherited from the clinically unaffected father, whereas the NM_000089.3(COL1A2):c.3473A>C (p.Asn1158Thr) variation was inherited from the clinically unaffected mother. Hence, homozygous likely pathogenic NM_018112.3(TMEM38B):c.112 + 1dupG variation was likely to be responsible for the disease in this patient.
Discussion
Molecular diagnosis of OI provides a basis for genetic counseling and allows possible prospective targeted therapies (Marini et al., 2017). Hence, it is very important to define the genetic background of this clinically heterogeneous disease. The growing list of genes responsible for the pathogenesis of OI increases the need for using advanced technologies like HTS to define the underlying causes in the patients. In this study, by using this massively parallel sequencing approach, we determined the genetic basis of 39 out of 43 OI patients taken from 5 different regions of Turkey and found 9 novel causative variants.
Pathogenic/likely pathogenic variations in COL1A1/COL1A2 genes are responsible for the majority of OI (Zhytnik et al., 2017). Glycine missense substitutions have been related to the more severe phenotypes in COL1A1-/COL1A2-related OI (Pack et al., 1989; Ohata et al., 2019). Phenotypic severity and diversity were higher among the patients with COL1A1/COL1A1 missense mutations compared to patients who have a splice site or premature termination mutation in COL1A1/COL1A2 genes. We suggest that rare missense variations with unknown clinical significance are challenging the diagnosis and genetic counseling of OI. COL1A1 and COL1A2 pathogenic variations are known as full penetrant (Steiner and Basel, 1993). However, some studies are suggesting otherwise, bringing forward an incomplete penetrance in this type of OI too (Pereira et al., 1994).
Segregation analysis is an important tool for classifying the variants of unknown significance in predominantly inherited diseases, such as COL1A1-/COL1A2-related OI (Garrett et al., 2016). Nevertheless, the reduced penetrance can complicate genetic counseling. We determined three different rare missense variations in our patient B12 and B40, complicating the genetic diagnosis. NM_000089.3(COL1A2):c.671G>A (p.Arg224His) variation was showing strong in silico predictions for pathogenic potential. However, when we consider both the de novo nature of the other variation in the patient, c.1040G>A in COL1A1, which causes a glycine to aspartic acid change in position 347, and with the supportive entry in the ClinVar database about the unknown significance of c.671G>A variation in COL1A2, we suggest that the c.1040G>A change in COL1A1 is the underlying cause of the disease.
Another missense variation, the NM_000088.3(COL1A1):c.77G>A (p.Gly26Asp) variation, defined in the patient B40 who also has a likely pathogenic c.112 + 1dupG variation in TMEM38B. Although the NM_000088.3(COL1A1):c.77G>A (p.Gly26Asp) variation is listed as a disease-causing mutation in HGMD database (Stenson et al., 2017), it was initially reported in a patient with another pathogenic COL1A1 variant whose OI phenotype is possible because of the second mutation. Our study also brings forward a case with a patient that was found to be inherited from an unaffected father (Fuccio et al., 2011). Together, these facts support the suggestion that all of the glycine missense substitutions are not equally damaging (Xiao et al., 2011) and genetic counseling should be given carefully. These findings also highlight the need for a combination of HTS and segregation analysis, in addition to silico assessments, for the interpretation of variants in clinically heterogeneous diseases.
Pathogenic/likely pathogenic variations of the FKBP10 gene were first found to be responsible for the Bruck Syndrome (OMIM) (Moravej et al., 2015). Alanay et al. (2010) reported that homozygous pathogenic variations of the FKBP10 gene were responsible for a severe autosomal recessive form of OI independent from Bruck Syndrome. c.890_897dup (p.Gly300Ter) variation in the FKBP10 gene has been reported to be associated both with arthrogryposis (Pehlivan et al., 2019) and with the OI type XI in ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000631496.2, accessed May 9, 2020). In our patient population, this variation has been found in two unrelated patients, and their phenotype was compatible with OI type XI. Our findings thus further support the studies on the role of pathogenic/likely pathogenic variations in the FKBP10 gene in OI type XI.
Defects in the P3H1 gene, previously known as LEPRE1, have been reported in the patients with OI type VIII, a severe recessive OI type (Cabral et al., 2007). In this study, we determined a heterozygous likely pathogenic NM_022356.3(P3H1):c.2055 + 18G>A variation in patient B32. The pathogenicity of this variation has been implicated in a study (Willaert et al., 2009). The inheritance pattern of P3H1-related OI is autosomal recessive. Therefore, likely pathogenic heterozygous NM_022356.3(P3H1):c.2055 + 18G>A variation in our patient B32 does not solely explain the P3H1-related OI in this patient. This patient also had a VUS in P3H1 and segregation analysis revealed that these two mutations were compound. Interestingly, a study shows that the heterozygous pathogenic variant in the P3H1 gene is associated with a moderate OI in a study (Santana et al., 2018). The pathogenic variation in this previous study was also compound heterozygous with a variant of unknown significance. Our findings support the suggestion that heterozygous pathogenic variations of P3H1 gene may be related to a moderate phenotype of OI type VIII. However, further functional studies for the variants of unknown significance that are compound heterozygous with a pathogenic variant in the P3H1 gene are strongly needed.
Homozygous or biallelic CRTAP gene defects were identified in patients with a distinct form of OI (Marini et al., 2010). This type of OI, labeled as OI type VII, has a severe-to-lethal osteochondrodysplasia with rhizomelia. neonatal fractures, broad undertubulated long bones, and fragile ribs (Marini et al., 2010). We found a homozygous novel likely pathogenic variation c.986delA (p.Lys329ArgfsTer36) in a 5-year-old female patient with perinatal fractures and growth retardation. This patient could not walk independently.
IFITM5 gene defects have been implicated in the diagnosis of OI type V. Almost all of the causative variant is a dominant mutation in the 5′UTR of IFITM5, creating a new start codon and elongating the N terminus of the bone-restricted interferon-induced transmembrane protein-like protein (BRIL; also known as IFM5) by five amino acids (Cho et al., 2012; Semler et al., 2012). Hyperplastic callus formation, one of the common features of OI type V, had been detected in two of our patients. Both of these patients were carrying the same likely pathogenic c.-14C>T variation in IFITM5 gene. Dentinogenesis imperfecta is not a common feature of OI type I; however, one of our patients with OI type V, dentinogenesis imperfecta, was identified.
TMEM38B gene defects have been implicated in recessively inherited OI type XIV (Rubinato et al., 2014). TMEM38B-related OI was determined in only one patient with a mild OI phenotype in our study population.
There are some restrictions and limitations of this study. First, we could not define the genetic background of 4 out of 43 patients. This may be due to use of a targeted gene panel. Performing genome sequencing may help to clarify the molecular defects, as the number of genes related to OI subtypes are increasing exponentially. The second limitation of this study is that we did not apply a functional study for novel variants, but instead used in silico prediction tools, databases, and segregation analysis for predicting the pathogenicity of the variants. A project is underway for the functional study of the novel variants found in our center's patient population.
In conclusion, we suggest that a multigene HTS approach is needed for the precise genetic diagnosis of OI in patients because of the large spectrum of genes that can be responsible for the disease. Although pathogenic variations of COL1A1 and COL1A2 genes are responsible from the majority of the OI phenotypes, these genes also harbor some rare variants with unknown significance, complicating the interpretation of genetic background of the patients. A multigene HTS approach, with the help of segregation analysis, is helpful for a discrete genetic diagnosis, preventing human geneticists from misinterpreting the rare variants that can be found simultaneously in the patients.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this study.