
Abstract
Background:
The association between dysregulated microRNAs (miRNAs) and acute myeloid leukemia (AML) is well known. However, our understanding of the regulatory role of miRNAs in the cytogenetically normal AML (CN-AML) subtype pathway is still poor. The current study integrated miRNA and mRNA profiles to explore novel miRNA-mRNA interactions that affect the regulatory patterns of de novo CN-AML.
Methods:
We utilized a multiplexed nanoString nCounter platform to profile both miRNAs and mRNAs using similar sets of patient samples (n = 24). Correlations were assessed, and an miRNA-mRNA network was constructed. The underlying biological functions of the mRNAs were predicted by gene enrichment. Finally, the interacting pairs were assessed using TargetScan and microT-CDS. We identified 637 significant negative correlations (false discovery rate <0.05).
Results:
Network analysis revealed a cluster of 12 miRNAs representing the majority of mRNA targets. Within the cluster, five miRNAs (miR-495-3p, miR-185-5p, let-7i-5p, miR-409-3p, and miR-127-3p) were posited to play a pivotal role in the regulation of CN-AML, as they are associated with the negative regulation of myeloid leukocyte differentiation, negative regulation of myeloid cell differentiation, and positive regulation of hematopoiesis.
Conclusion:
Three novel interactions in CN-AML were predicted as let-7i-5p:HOXA9, miR-495-3p:PIK3R1, and miR-495-3p:CDK6 may be responsible for regulating myeloid cell differentiation in CN-AML.
Introduction
The pathogenesis of acute myeloid leukemia (AML) is associated with the accumulation of acquired genetic mutations and epigenetic changes in hematopoietic progenitor cells that affect the proliferation, differentiation, and maturation of myeloid cells (Estey and Dohner, 2006). These changes manifest as abnormalities in the chromosomes. Thus, cytogenetics has emerged as a useful tool for AML patient risk stratification and has become one of the most important prognostic variables in treatment decisions (Trino et al., 2018). However, a sizeable number of patients are classified as having cytogenetically normal AML (CN-AML) and exhibit no chromosomal abnormalities (Estey and Dohner, 2006), limiting the usefulness of cytogenetics.
Therefore, in current practice, as updated by the recent 2016 World Health Organization classification, both cytogenetics and molecular diagnostics are utilized to narrow down AML subtypes to provide prognostic value and better treatment (Arber et al., 2016). Generally, CN-AML is categorized as posing intermediate risk, but that is not often the case, as patient outcome varies due to its high heterogeneity. Molecular prognostic biomarkers, which can help refine patient risk, offer a chance to fill in the gaps in AML knowledge. Success in prognosis and survival of AML patients has progressed in leaps and bounds for a cancer that was once a certain death sentence (Goldberg et al., 2018).
In the past decade, molecular abnormalities in AML have been highlighted after the discovery of several genetic mutations in CN-AML, especially those in FMS-like tyrosine kinase 3 (FLT3), nucleophosmin (NPM1), and CCAAT/enhancer-binding protein alpha (CEBPA) (Marcucci et al., 2011). One immediate result of this breakthrough can be seen in the prognostic evaluation of CN-AML patients. Screening for such mutations has become a common practice at the clinical level, and mutation identification may help to optimize therapeutic approaches in this group of patients. Surprisingly, there are many patients who do not have such mutations, such as some Malaysian patients (Mat Yusoff et al., 2019). These findings suggest that there are many more uncharacterized molecular biomarkers.
Evidence indicates that microRNAs (miRNA), short noncoding RNAs ∼22-nucleotides long, act as a causal factor for the pathogenesis of AML by regulating gene expression via translation repression and/or degradation of mRNA (Iorio and Croce, 2012). Dysregulation of miRNAs contributes to the pathogenesis of AML by reducing the differentiation of granulocytes and monocytes (Riccioni et al., 2015), activating p53 pathways that induce apoptosis in leukemic cells, inhibiting interferons, which in turn increase proliferation of hematopoietic stem and progenitor cells (Wallace et al., 2017), and inducing DNA hypomethylation as well as repressing tumor suppressing genes (Wong et al., 2019). However, the determination of miRNA function has proved to be rather difficult since miRNAs are estimated to regulate up to 30% of the human genome. In addition, a single miRNA is capable of targeting multiple genes and vice versa.
To address this problem, our study utilized the NanoString platform to measure miRNA and mRNA expression profiles from a similar set of samples and select the most suitable miRNA and mRNA targets. Possible interactions between these molecules were determined using a correlation study and then visualized through a network to identify interaction patterns. To narrow down the role of miRNA-mRNA interactions in CN-AML, gene ontology with functional annotation related to the disease was performed. Finally, we visualized these relationships using sequence- and conservation-based prediction algorithms from TargetScan.
miRNA-mRNA interaction studies in CN-AML are underreported compared to other AML subtypes. Experiments with large sample sizes, such as The Cancer Genome Atlas study (Network, 2013) reported that the majority of patients with AML were CN-AML (47%). The study also analyzed multiple AML subtypes to identify common mutations across AML. The present study is different because it focuses solely on CN-AML and fills the knowledge gaps pertaining to miRNA-mRNA interactions in CN-AML. Despite being categorized as posing intermediate risk, CN-AML may contain various mutations, each with different clinical outcomes (Marcucci et al., 2011). This study shows that CN-AML lacks proper risk stratification; thus, additional discovery studies to further understand CN-AML are justified.
Materials and Methods
Patients
Ethics approval from the Medical Research and Ethics Committee, Ministry of Health, and the University of Malaya Ethics Committee was obtained before commencement of the study (Ethics reference number: NMRR-16-1129-30996.) Written consent was obtained from all patients. Twenty-four newly diagnosed CN-AML patients (13 males and 11 females), with a median age of 43.5 years and age range of 18-72 years, were matched for age, sex, and ethnicity to controls at a 1:1 ratio (Table 1). Patients were mostly of Malay ethnicity (n = 15), followed by Chinese (n = 8) and Indian (n = 1), which portrays the racial diversity of Malaysia.
Patient Characteristics and Breakdown of Total Measured miRNA and mRNA
Patient blood samples were obtained from the Hospital Ampang, Selangor and Hospital Sultanah Aminah, Johor, while healthy controls were obtained from the University of Malaya Medical Center. Patients were recruited from November 2016 to April 2018. All patients exhibited blast percentages above 90%. Inclusion criteria for patients were early diagnosis of CN-AML and age older than 18 years. Exclusion criteria for patients were a history of medical interference, secondary AML, and AML other than CN-AML. These criteria were set to ensure the best de novo presentation of the disease. For control subjects, inclusion criteria were age older than 18 years, healthy (no history of medical intervention for the past 6 months and currently not diagnosed with any form of diseases), and underwent clinical screening.
Total RNA extraction
The buffy coat was separated from the peripheral blood using Ficoll-Paque density gradient media and lysed using QIAzol Lysis Reagent (Qiagen, Hilden, Germany). Total RNA was extracted using the miRNeasy Kit (Qiagen). RNA purity and integrity were assessed using a spectrophotometer and the Agilent RNA 6000 Nano Kit (Agilent, Waldbronn, Germany), respectively.
Experimental assays
The miRNA and mRNA profiles were obtained using the nCounter Human v3 miRNA expression assay kit and nCounter PanCancer Pathways Panel Kit (NanoString Technologies, Seattle, WA), following the manufacturer's protocols. The kits utilize complex color code probes, comprising four colors in six positions. This allows detection of large numbers of cancer-related miRNAs (n = 777) and mRNAs (n = 730) in a single sample. The product utilizes the direct ligation of oligonucleotide tags onto total RNA, thus allowing miRNA and mRNA to be directly measured with great specificity and sensitivity, while eliminating the use of reverse transcriptase or amplification. The microarray data were deposited in the Gene Expression Omnibus database (accession number GSE142700).
Data analysis
nSolver Analysis version 3.0, an in-house software program, was used to interpret the results obtained from the experimental assays. The software normalizes raw data generated from the NanoString microassays to calculate differential expression (fold change) and compares the case-control using the independent t-test with an adjusted p-value (false discovery rate, FDR). Significant miRNAs and mRNAs were selected, followed by the construction of an miRNA-mRNA correlation matrix using R, where the Pearson correlation was adopted with the adjusted p-value (FDR).
For a better understanding of the miRNA-mRNA interactions, the miRNA-mRNA correlations were visualized as a network using miRTarVis, a licence-free program running Java (Jung et al., 2015). Since a large number of miRNA target genes were identified, Gene Ontology (GO) analysis was performed using PANTHER to identify functional groups of genes. Finally, the miRNA-mRNA pairs in this experiment were submitted to TargetScan, a reputable miRNA target prediction database. Another target prediction database, microT-CDS was used for comparison.
For this study, we defined significant miRNA and mRNA expression as a p-value <0.05, with a fold change greater than 1.5 or less than 1.5 (FC >1.5 or FC < −1.5). The miRNA-mRNA pair correlations were set at r <−0.3. Since miRNA regulates mRNA expression, we were only interested in negative correlations. The cutoff r <−0.3 was defined as a weak negative correlation based on an article published by Mukaka et al. (Mukaka, 2012).
Results
Differential miRNA and mRNA expressions in CN-AML
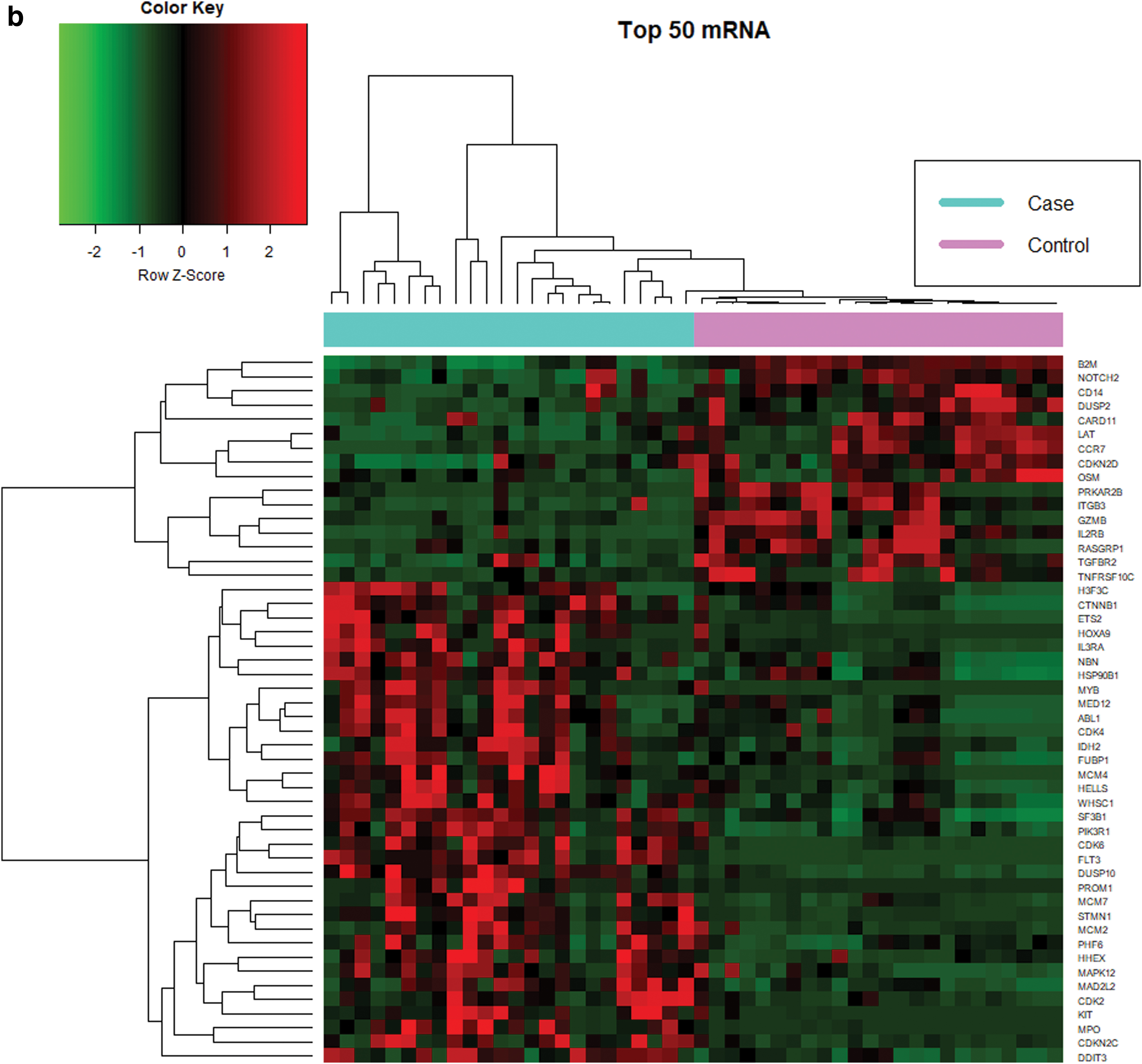
We profiled 777 human miRNAs in CN-AML patients. Significant differentially expressed miRNAs were called when the prerequisites of a fold change greater than 1.5 and an adjusted p < 0.05 were met. Based on these parameters, 53 significant differentially expressed miRNA signatures (39 downregulated and 14 upregulated, Table 1 and Supplementary Data S1) were identified. Figure 1a shows the heat map for the top 50 significant miRNAs. Notably, several signatures linked to CN-AML were identified, including let-7, miR-181, and miR-199.
We also observed several well-studied miRNAs in cancers, such as the miR-151 family, which has been reported in various cancers, such as breast cancer (Krell et al., 2012; Yeh et al., 2016), prostate cancer (Chiyomaru et al., 2012), gastric cancer (Hsu et al., 2016), esophageal cancer (Gu et al., 2013), and cholangiocarcinoma (McNally et al., 2013). The mir-151-5p family was also reported to be upregulated in AML, while its counterpart, mir-151-3p (or miR-151*), was upregulated in acute lymphoblastic leukemia (de Leeuw et al., 2014).
The expression of 730 human mRNAs was quantified in the same samples as the miRNA set. We observed 71 differentially expressed mRNA signatures (18 downregulated and 53 upregulated, Table 1 and Supplementary Data S2) under similar prerequisites. Figure 1b highlights the top 50 significant mRNAs. We also identified eight significant genes that are directly involved in the AML pathway (Fig. 2): FLT3, RUNX1, MPO, CD14, CEBPA, KIT, MYC, and PIK3R1.

Significant genes in the AML pathway. The figure illustrates the AML pathways, which would best represent the CN-AML pathway. Green box represents upregulation while red box represents downregulation. Genes within colored boxes are significant. AML, acute myeloid leukemia. Color images are available online.
Detection of miRNA-mRNA correlation and coexpression networks
To examine the functional correlation between miRNAs and mRNAs, we integrated both results to form a correlation matrix. The matrix yielded a total of 3763 miRNA-mRNA pairs. Given that miRNAs often negatively regulate gene expression, we hypothesized that negatively correlated miRNA-mRNA pairs should be enriched for target interactions. Based on this rule, we identified 636 significant pairs (r < −0.3) (Table 2 and Supplementary Data S3). A correlation network was constructed to visualize the correlation between the miRNAs and mRNAs using the Kamada-Kawai algorithm (KK-layout) (Fig. 3). This algorithm is a force-directed graph that focuses on centrality and is suitable for identifying communities within a network.
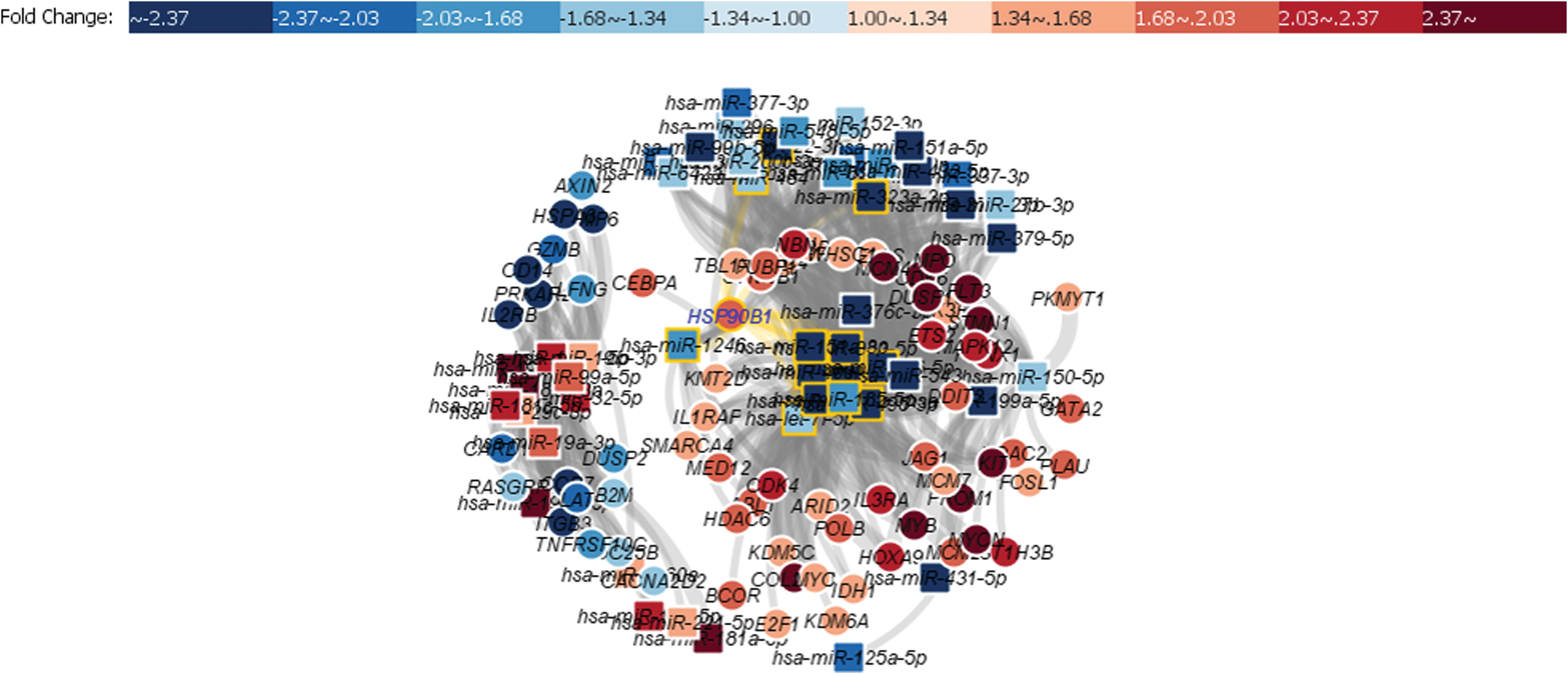
Network of 637 significant miRNA-mRNA correlations. Squares are miRNAs while circles are mRNAs. Red color indicates upregulation while blue color indicates downregulation. The center of the network indicates a group of miRNAs with multiple target mRNAs. Color images are available online.
Break Down of Total miRNA-mRNA Correlation
In this network, we identified a cluster of 12 miRNA signatures that were centrally located. These miRNAs are responsible for regulating 73% of the identified mRNAs; thus, we speculate that these miRNAs could contribute to CN-AML. Within this cluster, five miRNAs, miR-495-3p, miR-185-5p, let-7i-5p, miR-409-3p, and miR-127-3p, presented with the most mRNA targets; 38, 34, 34, 30, and 30 targets, respectively, signifying their importance.
Functional enrichment analysis of CN-AML patients
Our subsequent analysis investigated the functional impact of the target genes of our five miRNAs of interest: miR-495-3p, miR-185-5p, let-7i-5p, miR-409-3p, and miR-127-3p. Using PANTHER (The Gene Ontology Consortium, 2016; Mi et al., 2018), we identified the GO biological processes of miRNAs and their respective target genes (Table 3). These five miRNAs have been reported to play vital roles in the dysregulation of hematopoiesis, which is an important process in cancer development (Hanahan and Weinberg, 2000).
Gene Ontology Analysis of miRNAs of Interest
GO, gene ontology; FDR, false discovery rate.
We observed that miR-495-3p, miR-185-5p, and miR-127-3p were associated with the same biological processes: negative regulation of myeloid leukocyte differentiation, negative regulation of myeloid cell differentiation, and positive regulation of hematopoiesis. Furthermore, all three miRNAs targeted almost the same group of genes with only small differences. We hypothesized that these three miRNAs were involved in a similar pathway for myeloid leukocyte differentiation. Let-7i-was also associated with myeloid cell differentiation; however, with different gene sets from the three miRNAs. This indicates its involvement in a different pathway. Finally, GO analysis showed that miR-409-3p was involved in the regulation of hematopoiesis.
Predicted miRNA-mRNA interaction
We further determined whether the miRNAs regulate the predicted targets. We performed a computational analysis to predict possible matches between the 3′-UTR of the mRNAs and seed sites of the miRNAs. We submitted the five selected miRNAs and their target genes involved in hematopoiesis to TargetScan. To ensure a high prediction rate, we only considered pairs that were canonical. Table 4 summarizes the predicted sites of miRNA-mRNA binding. From the results, only three interactions were predicted: let-7i-5p:HOXA9, miR-495-3p:CDK6, and miR-495-3p:PIK3R1. These interactions were reproducible (miTG score 0.87, 0.99, and 0.87, respectively) in the microT-CDS prediction databases.
Predicted miRNA-mRNA Interactions
Let-7i-5p was predicted to regulate HOXA9 at three possible target positions: 1409-1415, 1084-1090, and 194-200 in the HOXA9 3'UTR. For miR-495-3p, TargetScan predicted the miRNA to regulate PIK3R1 and CDK6. MiR-495-3p targets the PIK3R1 3′UTR at three possible positions: 914-920, 930-936, and 1510-1516. miR-495-3p was also predicted to target CDK6 at seven positions, the maximum number of target positions per gene. This suggests that miR-495-3p heavily regulates CDK6 genes.
Knowing that the mutational status of CN-AML, such as FLT3 and NPM1, can contribute to CN-AML prognosis, we further looked at these interactions stratified by patients with FLT and NPM1 mutations. There were six (25%) CN-AML patients with FLT3-ITD mutations, five (20%) with NPM1 mutations, and three (12.5%) with both mutations. All three interactions remained significant (p < 0.05) in any of these subgroups except for miR-495-3p:CDK6 in patients with both mutations (Supplementary Table S1). Let-7i-5p:HOXA9 had the strongest correlation in CN-AML patients with only +FLT3-ITD, only +NPM1, and both +FLT3-ITD and +NPM1 (r = −0.70, −0.82, and −0.91, respectively).
Discussion
The field of miRNA biology has expanded considerably over the past decade. The role of miRNAs in cancer development is unquestionable and has been comprehensively discussed (Ding et al., 2018; Dufresne et al., 2018). Based on the notion that a single miRNA can target multiple mRNAs and vice versa (Li et al., 2019), the miRNA-mRNA regulatory relationship generates a vast amount of complex data, presenting a formidable challenge for researchers to elucidate the role of each gene.
Advancements in profiling techniques with the aid of bioinformatics have enabled us to analyze large numbers of miRNA and mRNA datasets in a single test. In this study, we developed an experiment-based functional miRNA-mRNA dataset and illustrated how these data can be used to create an miRNA-mRNA interaction network within a biological context, enabling the discovery of functional biological pathways involved in the development of CN-AML.
Integrated analysis revealed a cluster of 12 miRNA signatures within a network representing 637 constructed pairs that are involved with a variety of genes implicated in the pathogenesis of AML. The study identified five putative miRNAs (miR-495-3p, miR-185-5p, let-7i-5p, miR-409-3p, and miR-127-3p) that play pivotal roles in gene regulation in CN-AML. Further analysis by TargetScan revealed three miRNA-mRNA interactions in CN-AML, let-7i-5p:HOXA9, miR-495-3p:PIK3R1, and miR-495-3p:CDK6, which had previously not been reported.
Little is known about the functional role of the let-7 family in normal human physiology. However, a number of reports have emerged on its close association with cancers. Let-7 has a developmental role, and this was evidenced by the significant reduction of differentiation and proliferation of embryonic cells by the blockade of let-7 by LIN28 (Viswanathan et al., 2008). Interestingly, one study also reported that an increase in LIN28B, a variant of LIN28, through the activation of PRL-3, results in the induction of a stem cell-like transcriptional program in AML, thus contributing to its leukemogenesis (Zhou et al., 2017b).
Zhou et al. (2017a) further demonstrated that inhibition of LIN28B impairs the growth of leukemic cells by targeting LIN28B/let-7/IGF2BP1. The involvement of let-7 in the embryonic stage also explains early progression of cancers by targeting the embryonic gene HMGA2, in line with the hypothesis that cancers are cases of reverse embryogenesis (Park et al., 2007; Barh et al., 2010). In a blast crisis chronic myeloid leukemia (CML) mouse model, impaired let-7, in addition to the upregulation of LIN28, led to increased leukemia stem cell generation (Zipeto et al., 2016).
Our study outcome suggests that let-7i-5p can regulate hematopoiesis and differentiation of hematopoietic stem cells (HSCs) through the regulation of HOXA9. A separate study also suggested the possibility of a let-7i-5p interaction with HOX genes (Yoon et al., 2015). The HOX gene comprises four clusters, HOXA/B/C/D, and is generally responsible for the promotion of pathogenesis of acute leukemia and the self-renewal ability of leukemia stem cells, although HOXA and HOXB appear to be the key transcriptional regulators in hematopoiesis (Iacovino et al., 2009). Overexpression of HOXA9 causes expansion of HSCs and early progenitors, leading to myeloproliferative phenotypes in mice (Kroon et al., 1998; Thorsteinsdottir et al., 2002).
Myeloproliferation is unlikely to progress into full-blown AML on its own without other stimulating factors. For example, HOXA9 collaborates with MEIS1A in the progression to full-blown leukemia (Kroon et al., 1998; Collins and Hess, 2016). Thus, we speculate that the let-7i-5p:HOXA9 interaction may serve as a precursor for CN-AML progression. Dysregulation of let-7i has also been reported in other types of cancers, such as hepatocellular carcinoma (Yang et al., 2019) and cervical cancer (Chhabra, 2018).
In healthy tissues, miR-495-3p is involved in the development of stem cells, cartilage, pancreatic acinar cells, HSC differentiation, and neovascularization (Chen et al., 2017). MiR-495-3p was observed to have decreased expression in early myelopoiesis; however, there was a marked increase in a small set of AML (Cattaneo et al., 2015). In mixed-lineage leukemia (MLL)-rearranged AML, the downregulation of miR-495 was reported to greatly inhibit cell viability and increase cell apoptosis (Jiang et al., 2012). Here, we identified two gene targets of miR-495-3p, PIK3R1, and CDK6, which are associated with the pathogenesis of CN-AML.
PIK3R1 is a member of a family of lipid kinases. PI3K plays a key regulatory role in many cellular processes, including cell survival, proliferation, and differentiation, and is thus broadly associated with many types of cancer (Liu et al., 2009). The interaction between miR-495 and PIK3R1 is mostly found in hard tumors, such as breast cancer (Mishra et al., 2015) and endometrial cancer (Tan et al., 2019). The interaction between miR-495-3p and PIK3R1 has not been explored in leukemic patients as of today (the day of writing of this article). However, in AML, PI3K is directly involved in the spontaneous proliferation of primary AML cells through the PI3K-AKT signaling pathway (Kubota et al., 2004).
The inhibition of PI3K was observed to reduce cell survival of leukemic cells in AML (Xu et al., 2003). The upregulation of the PI3K-AKT-mTOR signaling pathway might also occur, possibly due to the oncogenic mutations of known AML-critical genes, including tyrosine kinase receptors, such as FLT3, cKIT, and BCR-ABL, or mutations in downstream factors, such as JAK, AKT, PTEN, and mTOR (Hoang et al., 2012). Further in vitro studies are required to test the effect of miR-495:PIK3R1 interaction on the PI3K-AKT signaling pathway and understand how it contributes to the progression of AML.
CDK6 is an important regulator of hematopoiesis by interfering with many genes and their pathways. CDK6 expression is responsible for the increased proliferation of myeloid progenitors, which is achieved by interference with AML-RUNX1 DNA binding, RUNX1-CEBPA interaction, regulation of FLT3, regulation of PIM1, and regulation of EGR1 (Fujimoto et al., 2007; Scheicher et al., 2015; Uras et al., 2016). CDK6 is also reported to be a key component in myeloid differentiation of AML with MLL-fusion, notably MLL-AF9 (Placke et al., 2014), and fits with miR-495 dysregulation in MLL rearrangements (Jiang et al., 2012).
Note that another form of MLL rearrangement, MLL-PTD, exists and affects 10% of CN-AML patients (Caligiuri et al., 1998). Judging by how both MLL fusions and MLL-PTD disrupt normal MLL function in maintaining epigenetic transcriptional memory at HOX gene loci (Zhang et al., 2012), there is a notion that a complex pathway exists between miR-495, CDK6, MLL, and HOX.
In this study, five miRNAs, miR-495-3p, miR-185-5p, let-7i-5p, miR-409-3p, and miR-127-3p, presented the most mRNA targets. Unfortunately, the TargetScan and microT-CDS databases only predicted let-7i-5p:HOXA9, miR-495-3p:PIK3R1, and miR-495-3p:CDK6 to be valid. The miRNAs that were not considered valid in this study, miR-185-5p, miR-409-3p, and miR-127-3p have been previously reported in other cancers. MiR-185 was demonstrated to regulate cell proliferation and glycolysis in AML cells in vitro (Zhang et al., 2020). A 4-miRNA scoring system developed by Zhu et al. ranked miR-185 as highly associated with overall survival (Zhu et al., 2019). In CML, miR-185 was downregulated and found to be a tumor suppressor (Fu et al., 2016).
A study reported the extent of miR-185 tumor suppressing capabilities by targeting PAK6, which increases the sensitivity of CML cells toward tyrosine kinase inhibitors (Lin et al., 2020). In addition, miR-409-3p was reported to be downregulated in AML with a high risk of relapse (Díaz-Beyá et al., 2014). In CN-AML, upregulation of DNMT3B was reported in various genes responsible for differentiation, proliferation, and survival pathways by interacting with miR-409-3p, resulting in adverse clinical outcomes (Niederwieser et al., 2015). Finally, miR-127-3p has been reported in various cancers, such as hepatocellular carcinoma (Yang et al., 2013) and colorectal cancer (Zhang et al., 2019). In leukemia, miR-127-3p is downregulated in myelodysplastic syndrome (Merkerova et al., 2015).
Reports on miRNA-mRNA relationships using an actual experiment-based model are scarce and, further, limited to the use of a similar set of patient samples for both profiles. Computational methods, such as TargetScan and microT-CDS, which were utilized in this study, were developed to quickly identify potential mRNA targets with limited empirical evidence for a small selection of miRNAs. In addition, the prediction algorithm poses a great challenge in yielding false positive results (Riffo-Campos et al., 2016). Here, we utilized both experimental and computational methods to address this challenge. Studies that integrate large miRNA and mRNA profiling data help researchers to view the relationship patterns of the extremely complex biological mechanisms of disease. A more refined analysis and scoring system for the diagnosis of AML is slowly being integrated into clinical settings (Chuang et al., 2015).
However, existing AML studies on miRNA-mRNA interactions only assessed general AML, without taking into account the heterogeneity of the disease (Cancer Genome Atlas Research Network, 2013; Gadewal et al., 2020). In this study, we strictly focused on CN-AML, an intermediate-risk AML subgroup with no karyotype abnormalities. We utilized the nCounter platform in this experiment, as it offers key advantages, such as the absence of an amplification step and direct measurement of target molecules, to avoid any bias. NanoString also offers several other key advantages, including sensitivity, technical robustness, and utility for clinical applications (Veldman-Jones et al., 2015).
Conclusion
Overall, these results open new perspectives toward understanding the molecular interactions and mechanisms associated with the pathogenesis of CN-AML. We identified three miRNA-mRNA interactions that could be associated with the regulation of myeloid cell differentiation in CN-AML: let-7i-5p:HOXA9, miR-495-3p:PIK3R1, and miR-495-3p:CDK6. However, functional studies need to be carried out to validate these findings.
Footnotes
Acknowledgments
We express our appreciation to Director General of Health, Ministry of Health, Malaysia for his permission to publish this article. We thank the Deputy Director General of Health (Research and Technical Support, Ministry of Health) and Director of Institute for Medical Research (IMR), Malaysia, for their continuous supports. The authors gratefully acknowledge the subjects for their participation in this study, investigators and staff of the IMR, UMMC, Hospital Ampang, and Hospital Sultanah Aminah.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by Fundamental Research Grant Scheme FP027-2018A.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.