
Abstract
Background:
Multiple osteochondromas (MO) are an autosomal-dominant disease characterized by the growth of multiple cartilage-capped prominences in the growth plate region of the metaphysis in long and flat bones.
Materials and Methods:
To detect genetic mutations related to MO, a three-generation Chinese family with MO was evaluated using whole exome sequencing for mutation screening. The candidate pathogenic mutation was validated by Sanger sequencing.
Results:
A novel frameshift (NM_000401.3:c.1321del:p.Leu441TrpfsTer28) in exon 8 of the exotosin 2 (EXT2) gene was identified in two affected individuals. Codons 441 and 468 in the EXT2 gene are highly conserved among vertebrates as demonstrated by multiple sequence alignment. The c.1321 del C resulted in an amino acid change at codon 441, which generated a premature stop codon at position 468, causing complete loss of the glycosyltransferase domain.
Conclusions:
A novel frameshift mutation c.1321delC detected in the EXT2 gene may help in prenatal genetic screening and early diagnosis of MO.
Introduction
Intrinsic to their names, cartilaginous tumors are characterized by containing cells with a morphology that resembles that of chondrocytes and can produce chondroid matrix; these tumors include osteochondroma, chondroma, chondroblastoma, and chondromyxoid fibroma. Multiple osteochondromas (MO), formerly known as hereditary multiple exostoses, are an autosomal-dominant disease with an incidence of ∼1:50,000 (Schmale et al., 1994). Penetrance occurs in ∼96% of females and 100% of males with MO (Schmale et al., 2013). In general, MO present as a benign tumor; however, 1-2% of patients with MO progress to osteosarcoma or chondrosarcoma (Jennes et al., 2009).
MO typically occur during childhood and continue to progress until the end of puberty. Its onset initiates with the formation of ectopic growth plate-like cartilage, which grows outward and forms exostosis (Jones, 2011). Ossification occurs at the proximal portion, whereas the distal portion may maintain the cartilaginous cap. The thickness of the cartilaginous cap often decreases with age. Compared with normal cartilage, the trabeculae in the cap are irregularly arranged and may contain calcified tissue. A reduction in skeletal growth, bone deformity, restricted joint motion, and shortened stature may occur in patients with MO. Expanding tumors may further compress the surrounding muscles, blood vessels, and nerves to cause pain, and surgery is currently the main treatment for this disease.
Although the molecular mechanism of MO is not completely clear, EXT1 and exotosin 2 (EXT2) are considered to be involved in the genesis of MO (Wuyts and Van Hul, 2000; Lonie et al., 2006). EXT genes encode proteins involved in the biosynthesis of heparan sulfate (HS), which plays an important role in chondrocyte differentiation, ossification, and apoptosis. HS has the ability to interact with proteins in some key signaling pathways, such as fibroblast growth factor (FGF), bone morphogenetic protein (BMP), and hedgehog protein family members (Billings and Pacifici, 2015). HS deficiency increases the expression of Indian hedgehog and boosts BMP signaling while dampening FGF signaling (Stickens et al., 2005; Ornitz and Marie, 2015; Dierker et al., 2016; Salazar et al., 2016). Zak et al. (2011) showed that symptom severity is negatively correlated with HS levels.
MO are associated with mutations in EXT1 (OMIN*608177) and EXT2 (OMIM*608210), which are located at 8q24 and 11p11-p12, and ∼80% of these mutations lead to loss of protein function. According to a previous study, patients with mutations in EXT1 are prone to having a larger number of exostoses and more bone-related abnormalities than those with EXT2 mutations, suggesting that EXT1 plays a more important role in the composition of HS (Jennes et al., 2009; Zuntini et al., 2010). Furthermore, mutations in EXT1 are more common than those in EXT2.
However, previous studies found that EXT2 mutations are more frequent in Chinese people (Kang et al., 2013; Guo et al., 2014). Interestingly, EXT2 mutations are only found in MO but not in sporadic osteochondromas. Some scholars have detected atypical chondroid tumors in MO, such as enchondroma, highlighting the roles of other disease-causing genes (Goud et al., 2015).
In this study, whole exome sequencing (WES) was performed on two patients from a three-generation Chinese family with MO, and the results were verified using Sanger sequencing. After comparing the data from the 1000 human genome data set, Genome Aggregation Database data set (Genome AD), and Exome Aggregation Consortium data set (ExAC), we identified a novel frameshift mutation as a possible pathogenic mutation, c.1321delC, in exon 8 of EXT2 that induces EXT2 protein truncation. The synthesis of HS is affected by damage in EXT2 protein function, which may cause MO.
Materials and Methods
Human subjects
The proband with MO was admitted to Zhejiang Provincial People's Hospital in 2020, and clinical diagnosis was performed based on accurate family histories and radiographical evaluation. The proband had a raised mass at the distal end of the femur at the age of 3 years; however, no medical examination or treatment was sought. We investigated the three-generation family and produced a family tree (Fig. 1). The proband's father (II-1) and grandmother (I-2) had lesions detected before age 11 years, and their heights were 172 and 160 cm, respectively. The proband underwent operation during this hospitalization because of an enlarged bilateral radius osteochondroma, whereas the proband's father and grandmother had not undergone surgery.
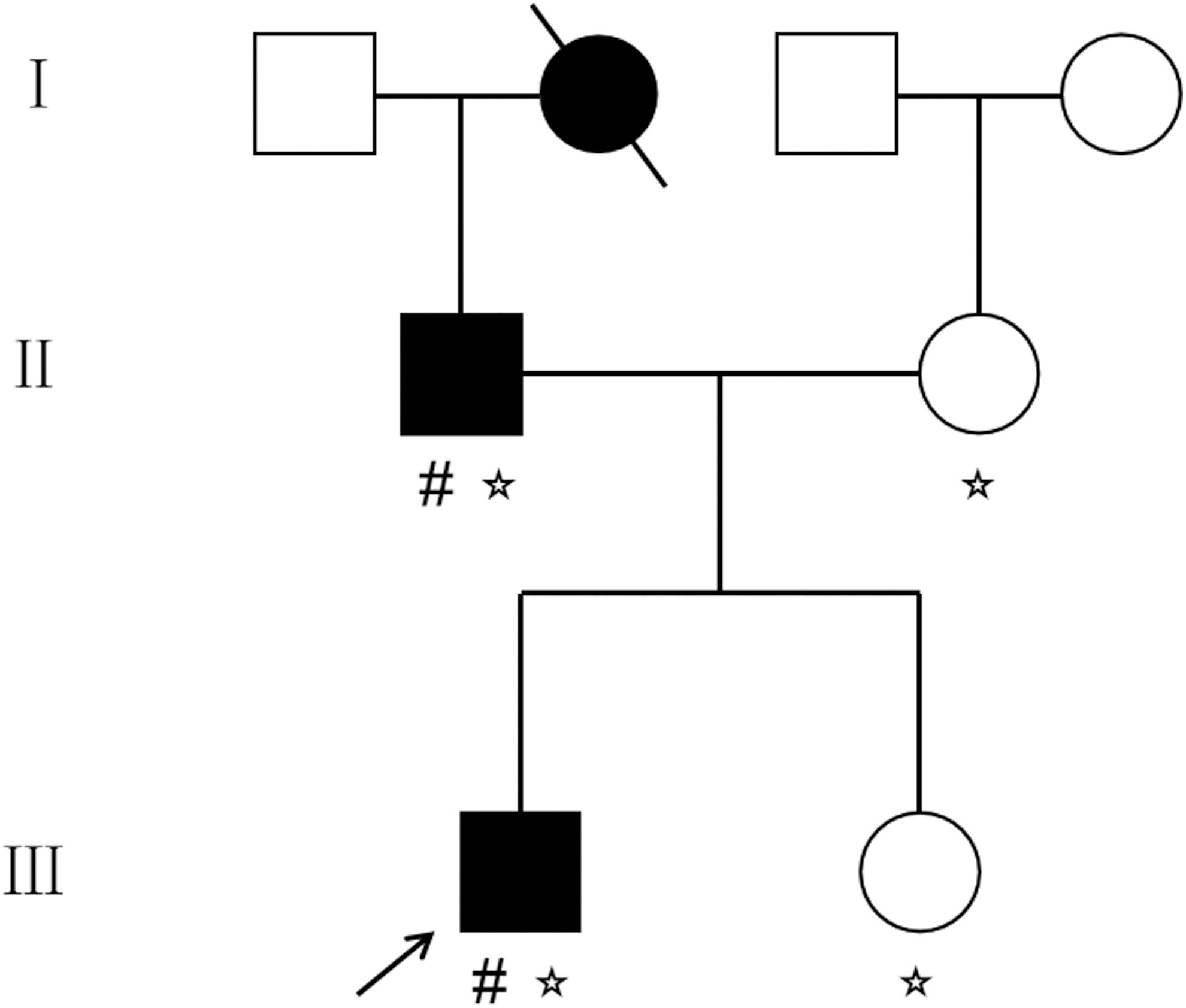
Pedigree of family with MO investigated in this study. The proband is indicated by a black arrow. (# represents individuals participating in WES. ☆ represents individuals participating in Sanger sequencing). MO, multiple osteochondromas; WES, whole exome sequencing.
This study was reviewed and approved by the ethics committee of the Provincial People's Hospital, and informed consent was obtained from all study participants.
Whole exome sequencing
Genomic DNA was extracted from peripheral blood samples of II-1 and III-1 according to the manufacturer's standard procedure (QIAamp DNA Blood Midi kit; Qiagen GmbH, Hilden, Germany). The genomic DNA was fragmented into 200-250 bp fragments with Bioruptor Pico to generate paired-end libraries. The library was enriched by array hybridization according to the manufacturer's instructions, followed by elution and postcapture amplification. The products were subjected to Bioptic Qsep 100 and ABI StepOne analyses to estimate the magnitude of enrichment. Finally, captured library sequencing was carried out on an Illumina HiSeq X Ten Analyzers (Illumina, San Diego, CA). Raw data were generated using the Illumina Pipeline software.
Sanger sequencing
We designed primers for standard polymerase chain reaction (PCR) assays using Primer Premier 5 software (Premier Biosoft, Palo Alto, CA) (Table 1). We designed primers for standard PCR assays using Primer Premier 5 software (Premier Biosoft) (Table 1). We performed Sanger sequencing on genomic DNA from blood samples of II-1, II-2, III-1, and III2. Mutation Surveyor software and Chromas (Technelysium Pty, Ltd., South Brisbane, Australia) were used to analyze the results by comparison with reference sequences from NCBI (NM_000401.3 for EXT2).
Primers Used for Sanger Sequencing the Exons of the EXT1 and EXT2 Genes
EXT, exostosin glycosyltransferase.
Variant analysis
WES reads were aligned with the human genome reference (hg19) using the Burrows Wheeler Aligner software tool (Li and Durbin, 2010). The output files were used to analyze sequencing coverage and depth. Single nucleotide variations (SNVs) and insertions-deletions (indels) were detected with Genome Analysis Toolkit software. All SNVs and indels were filtered and estimated using the 1000 human genome data set, Genome AD, and ExAC.
We predicted the effect of missense variants through dbNSFP, a database designed to facilitate this step by providing deleteriousness prediction and functional annotation for all potential nonsynonymous and splice-site SNVs in the human genome (http://database.liulab.science/dbNSFP), and screened mutations reported in published studies in the Human Gene Mutation Database and Clinvar Database. Pathogenic variants were assessed using American College of Medical Genetics and Genomics guidelines (Richards et al., 2015). Structural and functional changes in the EXT proteins caused by pathogenic mutations were analyzed using Mutation Taster.
Results
Patients and clinical imaging examination
In this family, the proband (III-1) was a 16-year-old boy. His father (II-1) and deceased grandmother (I-2) suffered from the same disease. X-ray examination revealed prominent osteochondromas in the proband's arms and knees. Exogenous osteochondromas occurred in the distal end of the bilateral radius and bilateral femurs, as well as the proximal end of the bilateral tibias (Fig. 2), manifesting as an irregular osteochondroma of varying size.
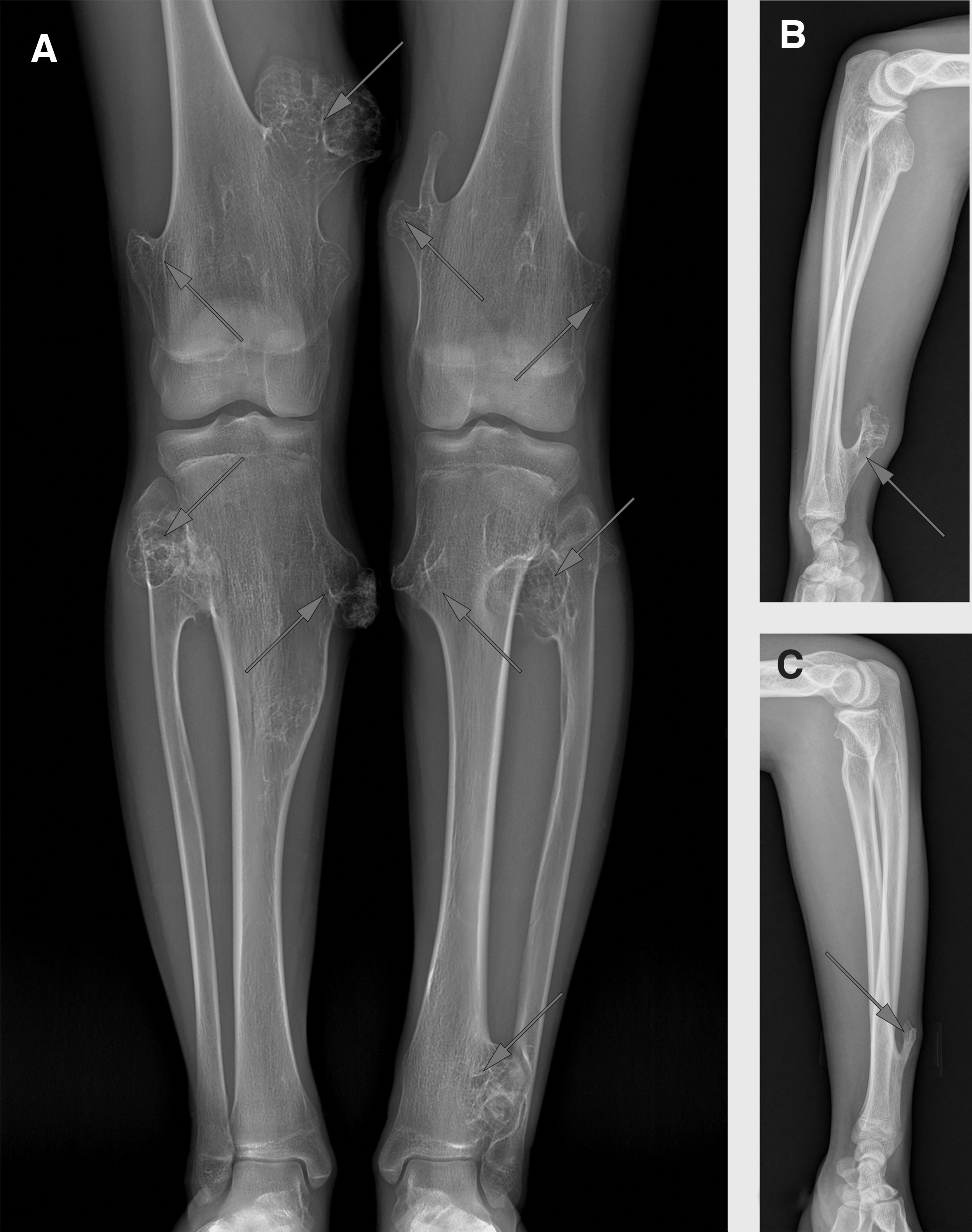
Clinical characteristics of the proband with MO.
WES and Sanger sequencing
The results of WES revealed 13,410 indels and 79,234 SNVs in III-1 and 13,240 indels and 77,279 SNVs in II-1. II-1 and III-1 were found to contain 8049 coexisting indels and 77,279 coexisting SNVs. Considering the importance of the functions of EXT1 and EXT2 in MO, one indel and five SNVs in both patients were screened out in EXT1 and EXT2. A frameshift mutation (NM_000401.3:c.1321del:p.Leu441TrpfsTer28) in EXT2 was regarded as a candidate pathogenic mutation considering the change in gene function caused by the mutation. The record of this mutation does not appear in 1000 human genome data set, Genome AD, and ExAC. Similarly, this mutation has not been reported in Leiden Open Variation Database. So this mutation ought to be confirmed novel.
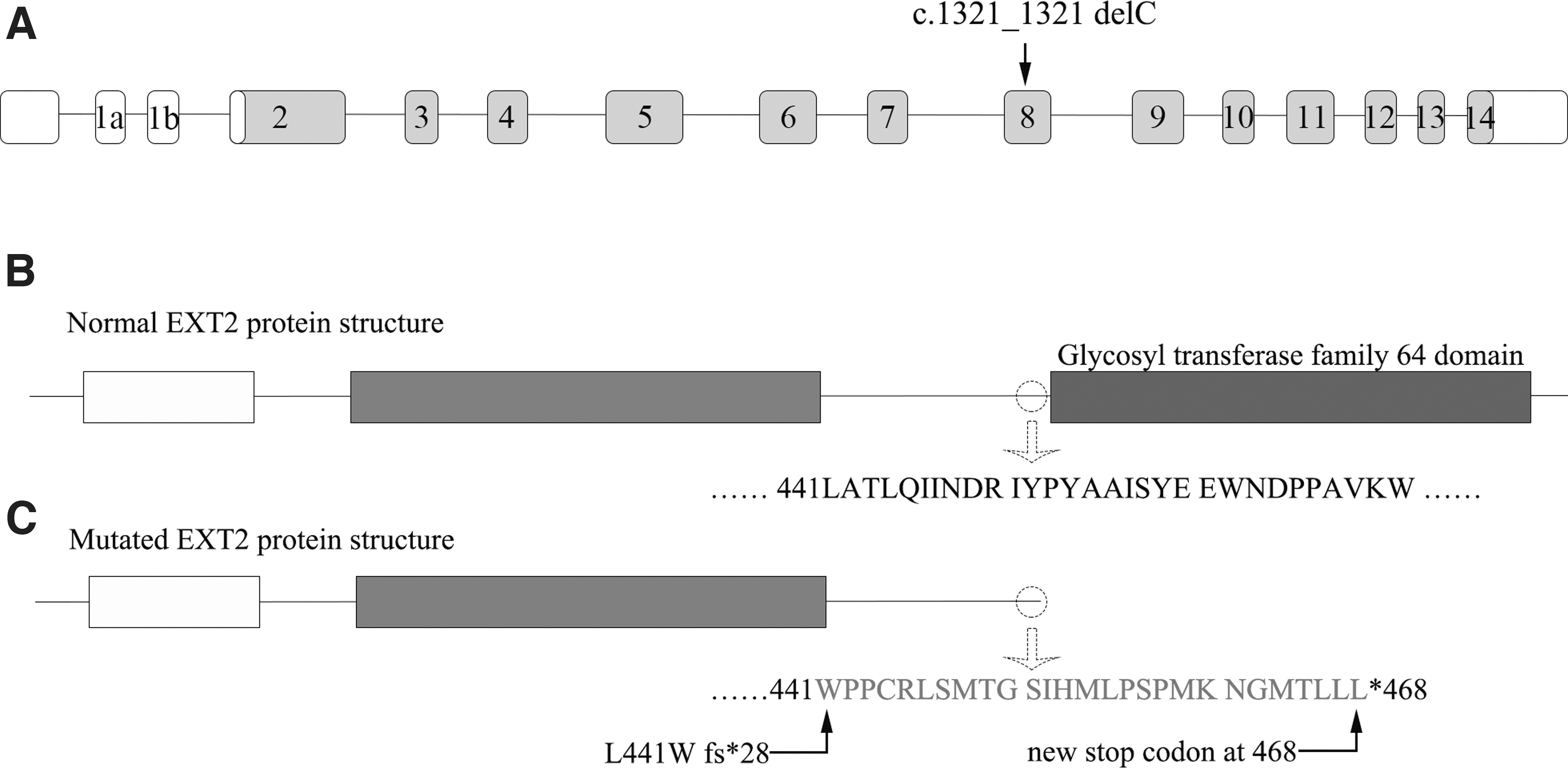
We verified this mutation using Sanger sequencing. The heterozygous deletion (c.1321 del C) in exon 8 of EXT2 was present in proband (III-1) and his father (II-1) but absent from his unaffected mother (II-2) and sister (III-2) (Fig. 3). The c.1321 del C is a one-nucleotide deletion resulting in an amino acid change at codon 441 to generate a premature stop codon at 468, causing complete loss of the glycosyltransferase domain (Fig. 4). Multiple sequence alignment showed that codon 441 of EXT2 was highly conserved among various vertebrate species (Fig. 5).
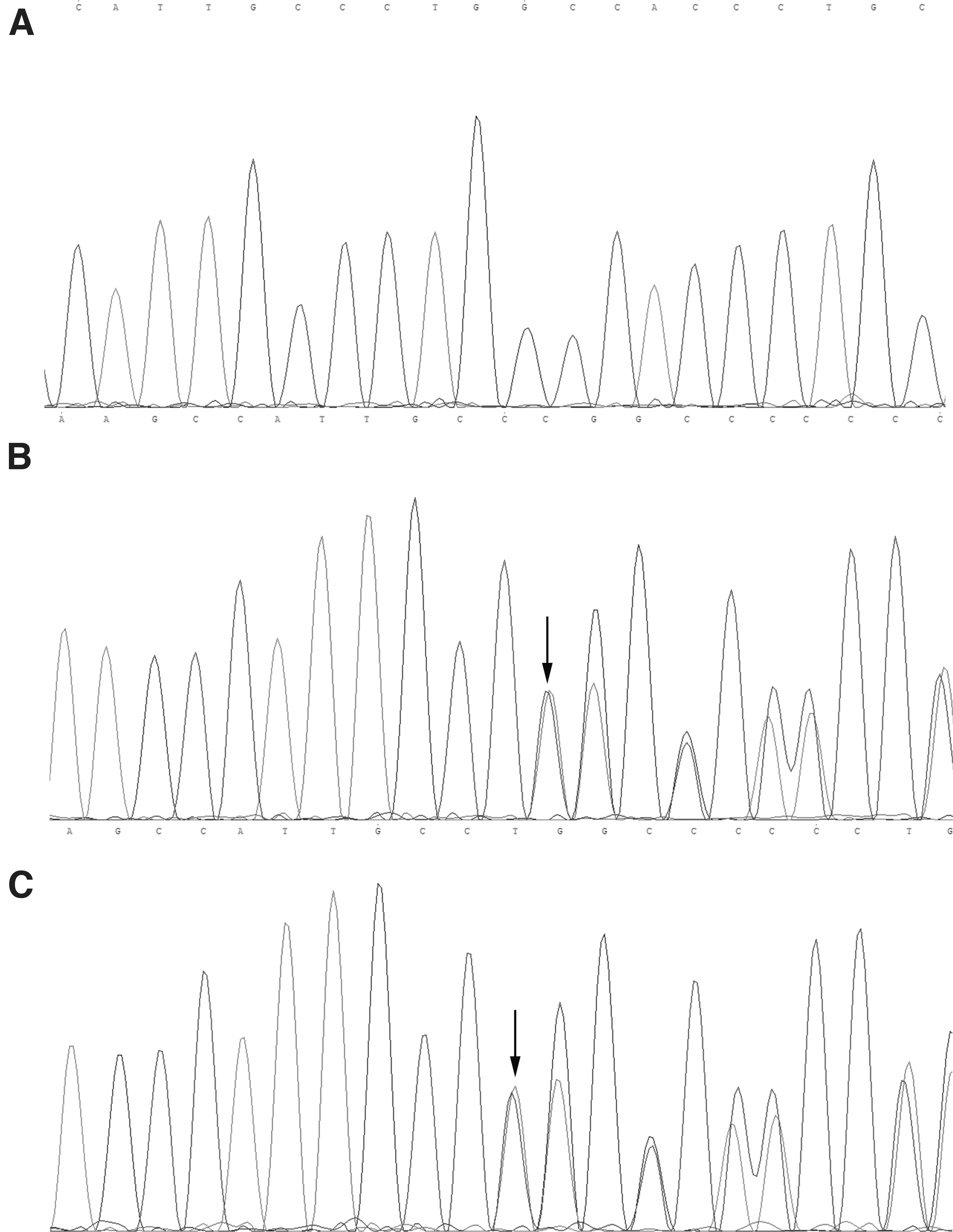
Novel frameshift mutation in codon 1321 of EXT2.

Multiple alignment between various vertebrate species.
Discussion
The common mutations found in EXT1 and EXT2 include missense, nonsense, splice-site, and frameshift mutations (Wuyts and Van Hul, 2000). However, previous studies revealed that a genomic rearrangement involving the first intron of EXT1 and a proximal noncoding DNA sequence has been detected in a family of MO patients who do not exhibit alterations in EXT exon or intronic splice donor sites (Waaijer et al., 2013).
Complementarily, another study found that an SAMD12-EXT1 fusion exists in a patient with osteochondroma, which partially disables the function of the EXT1 (Oliver et al., 2019). This finding indicated that in addition to exonic EXT alterations and intronic splice-site changes, gene fusion and genomic rearrangements within EXT1 introns are also a disease-causing mechanism of MO. Although neither of the above two conditions was found in this study, these mutations can be easily overlooked in the identification of MO mutations.
Li et al. (2018) found that single-base substitutions are more common in EXT2 mutations in Chinese patients. Brazil is the only country other than China that reports that EXT2 mutations are more common than EXT1 mutations. Santos et al. performed DNA sequencing (Sanger Method) and multiplex ligation-dependent probe amplification analysis on 114 families with MO. The results showed that 87.5% of EXT2 variants were located in regions encoding the exostosin domain, whereas 12.5% of EXT2 variants were located in regions encoding the middle segment of EXT2 gene, specifically in exon 8. No variants were found in the glycosyltransferase domain.
However, in EXT1 mutations, the same rate of EXT1 pathogenic variants is located in regions that encode the exostosin domain (45.2%) and transferase domain (45.2%) (Santos et al., 2018). Therefore, it is noteworthy that exon 8 is a high incidence region of mutations in EXT2.
In this study, a novel frameshift mutation in exon 8 of EXT2, c.1321 delC,was identified in two affected individuals from a three-generation Chinese family with MO. The c.1321 del C caused a change in amino acid change at codon 441, generating a premature stop codon at 468, resulting in the complete loss of the glycosyltransferase domain. As a result, the truncated protein lacked 284 amino acids compared with the normal EXT2 protein. The glycosyltransferase domain plays a crucial role in HS biosynthesis, and the damage to EXT2 protein function and dyssynthesis of HS may lead to MO. According to the distribution and number of the affected patients in this family suffering from MO, it can be inferred that c.1321 del C mutation originated from the father of the proband and was transmitted to the proband in an autosomal dominant manner.
The correlation between EXT2 mutation sites and clinical phenotypes remains unclear. The two patients in our study were an adult and a child, and both presented with multiple exostoses in the metaphysis. No symptoms of pain occurred in both patients, and there was no significant effect on axial bone growth. Compared with other family members, the height of the patients did not reduce significantly. The average adult height in this family is within the normal range of the Chinese adult height for age and gender.
Stickens et al. found that homozygous knockout of EXT2 in mice is embryonically lethal. Heterozygous EXT2 mice have a normal lifespan, with one-third of the mice developing one or more ectopic bone growths (Koziel et al., 2004). Our patients showed a heterozygous deletion in EXT2, supporting that the loss of a single allele is sufficient to cause MO.
In conclusion, the novel frameshift mutation c.1321 delC in EXT2 was identified in a three-generation Chinese family by exome sequencing and validated using Sanger sequencing. This mutation may be responsible for the disease pathogenesis in this three-generation family. Our current study extends the known mutation types of EXT2 and may facilitate prenatal genetic screening and early diagnosis of MO.
Footnotes
Authors' Contributions
Q.B. was involved in study design. Y.T. and J.L. were involved in data analysis and article writing. Y.T., J.L., Y.Z., Z.H., L.C., and Q.B. performed experiments.
Acknowledgments
We thank the patients and their families for their cooperation. Open access funding was provided by Zhejiang Provincial People's Hospital.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Science Foundation of China (Grant No. 81672769) and Natural Foundation of Zhejiang Province (Grant Nos. LQ18h060004 and GF19H060034).