
Abstract
Aim:
Our goal was to determine the genetic basis of early-onset myopathy in patients from two unrelated families.
Materials and Methods:
Whole-exome sequencing, autozygosity mapping, and confirmatory targeted Sanger sequencing were performed using genomic DNA extracted from blood samples from three myopathic patients of two unrelated families. Variant filtering and pathogenicity analyses were evaluated according to standard protocols and up-to-date pipelines applied at the King Faisal Specialist Hospital and Research Center.
Results:
A novel homozygous variant was detected in TTN gene within the first three M-line-encoding exons in a 9-year-old female in the first family who had delayed motor development and proximal weakness. Her 4-year-old affected brother, with the same homozygous variant, could not yet walk without help. This pathogenic nonsense variant is predicted to cause a premature stop during translation. In the second family we identified two novel variants as compound heterozygosites (a deletion and a variant affecting a canonical splice site) in an affected 9-year-old female with weakness that developed at age 3, in the second family. SpliceAI predicted the variants being splice-altering with high probability. These variants were fully segregated in the family. The deletion was found to be on the paternal allele, whereas the splicing variant was on the maternal allele. The patient's echocardiography revealed mitral valve prolapse with mild mitral regurgitation. Muscle histology showed minicores that were also confirmed by electron microscopy.
Conclusion:
Our study identified novel pathogenic variants in the TTN gene that are likely responsible for the phenotype of early-onset myopathy; hence, expanding genotype-phenotype relationship of titinopathies.
Introduction
The TTN gene (OMIM 188840) encodes titin, the largest known protein that is crucial for the structure, development, cell signaling, and elasticity of skeletal and cardiac muscle (Savarese et al., 2018a). Four distinct parts constitute the molecular architecture of titin, which spans half of the sarcomere. These are the N-terminal Z-disk, I-band, A band, and carboxyl-terminal M-line encoded by the last six exons of TTN (359-364, or Mex1 to Mex6) (Misaka et al., 2019).
Since the advent of next-generation sequencing, an expanding spectrum of cardiac and skeletal muscle diseases have been reported in both dominant and recessive TTN variants (Hackman et al., 2017; Savarese et al., 2018b). Such diseases are known as titinopathies (Savarese et al., 2020) and the affected individuals present with respiratory, cardiac, muscle problems, and infrequent facial weaknesses (Sasaki et al., 2020).
Early-onset recessive titinopathies include Salih myopathy (early-onset myopathy with fatal cardiomyopathy, OMIM 611705) (Hackman et al., 1993; Carmignac et al., 2007), congenital centronuclear myopathy (Ceyhan-Birsoy et al., 2013; Fattori et al., 2015), core myopathy with heart disease (Chauveau et al., 2014), childhood-juvenile-onset Emery-Dreifuss-like myopathy phenotype without cardiomyopathy (De Cid et al., 2015), and a congenital multicore titinopathy with fast myosin heavy chain deficiency (Perrin et al., 2020).
In this study, we describe two siblings and a third unrelated patient, with early-onset myopathy predicted to be due to pathogenic novel TTN variants. The first novel variant, a homozygous alteration detected in the two siblings within the first three M-line-encoding exons (Mex1, Mex2, and Mex3; exon 359, Mex 1) of TTN, known to cause Salih myopathy (OMIM 611705). Additionally, exome sequencing revealed two novel variants in compound heterozygositic state in the third patient.
Materials and Methods
Patient recruitment
We recruited three myopathic patients and unaffected members from two unrelated families from the central region of the Saudi Arabia for this study. The healthy parents from the first family are known to be consanguineous. There is no known consanguinity between the parents of the second family. The project was approved by King Faisal Specialist Hospital and Research Center IRB (RAC No. 2040042). The adult patients and their parents provided informed consent for the study.
Clinical descriptions
Patient 1 (Family 1)
Patient 1 is a 9-year-old girl born at term, after uneventful pregnancy with birth weight of 3.5 kg. She went through normal postnatal period. She was noticed to have right arm weakness by the age of 7 days attributed to Erb's palsy and improved with physical therapy. Her gross motor development was delayed. She sat unsupported by 8 months of age and walked by the age of 3 years. Currently, she is able to go upstairs and run but with difficulties. She is attending Grade 3 primary school with good performance. Her speech is appropriate for her age and she has normal hearing and vision. Her younger brother, patient 2, is similarly affected.
Physical examination (Table 1) revealed no dysmorphic features or ptosis, mild facial weakness, and high-arched palate. She had mild calf muscle pseudohypertrophy (Fig. 1A), decreased muscle tone (proximal>distal), and distal joint hyperlaxity (Fig. 1B). There was mild weakness of neck flexors (4+/5), proximal muscle weakness (4/5), lumbar lordosis (Fig. 1A), and waddling gait. She showed Gowers' sign when rising from the sitting position. She had spinal rigidity, flat chest associated with contracture deformity of the right elbow (60°), both knees (40°), and tight heel cords.
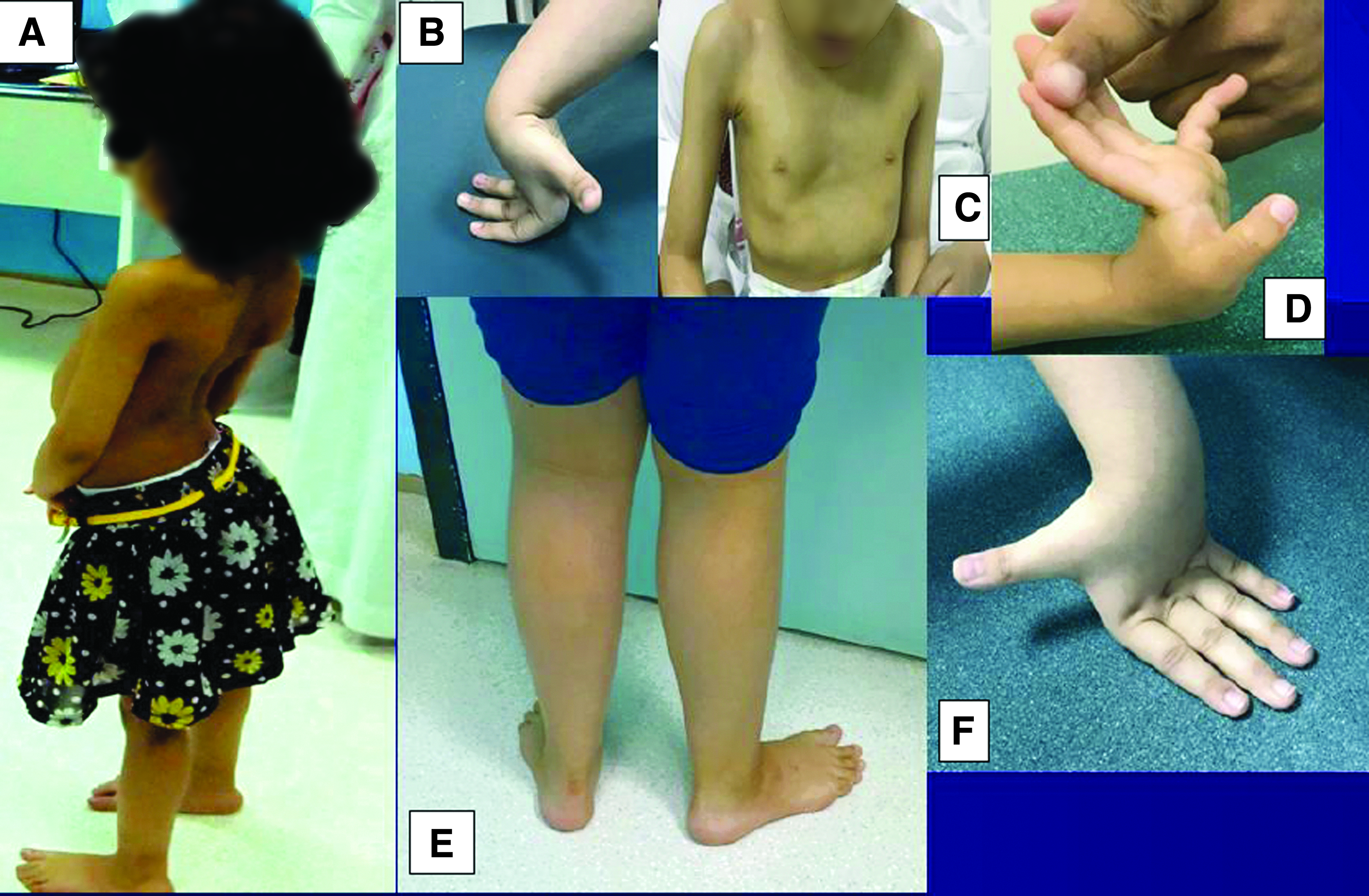
Clinical features of the three patients.
Clinical Features of Three Patients with Congenital Titinopathies
CK, creatine kinase; ECG, electrocardiography; F, female; M, male.
Electrocardiography (ECG, Supplementary Fig. S1) showed left axis deviation (left anterior fascicular block). Echocardiography revealed parachute-like mitral valve and persistent left superior vena cava to dilated coronary sinus, normal chamber size for body surface area, and good biventricular systolic function (Supplementary Fig. S2). Creatine kinase (CK) level was 886 U/L (N = 39-308).
Patient 2 (Family 1)
A 4-year-old boy, the younger sibling of Patient 1, was born at term through spontaneous vaginal delivery after uneventful pregnancy with birth weight of 2.7 kg. He went through normal postnatal period. He sat unsupported by 7 months of age, was able to crawl but could not yet walk without help. His speech is appropriate for his age and he has normal hearing and vision. Physical examination showed no dysmorphic features. There was decrease in muscle tone (proximal>distal), mild weakness of neck flexors (4+/5), and proximal muscle weakness (4/5). He had spinal rigidity, and deformed chest (pectus excavatum) associated with contracture deformity of both knees (20°). ECG (Supplementary Fig. S3) revealed left axis deviation (left anterior fascicular block). Echocardiography was normal and CK level was 911 U/L (N = 39-308).
Patient 3 (Family 2)
A 9-year-old girl was born at term to nonconsanguineous Saudi parents. Pregnancy history was significant for gestational diabetes, on dietary control. She was admitted to neonatal intensive care unit for 1 week for jaundice, which required phototherapy. She walked by the age of 14 months. Muscle weakness started gradually around 3 years of age when she developed difficulty in getting up from a sitting position. Currently, she is able to walk but can easily fall down. Her cognitive function is normal with good school performance.
Physical examination (Table 1) showed no dysmorphic features, high arched palate, mild calf pseudohypertrophy, and distal joint hyperlaxity (Fig. 1E, F). Manual muscle testing showed power of 4/5 at the shoulder, and pelvic girdles was 4/5, whereas the power of the distal muscles was normal. Deep tendon reflexes could not be elicited. She had spinal rigidity with mild scoliosis (concave to the left). Gowers' sign was positive and her gait was waddling.
ECG was normal and echocardiography revealed mitral valve prolapse with mild mitral regurgitation. Serum CK was normal (110 U/L, N = 26-192). T1-weighted magnetic resonance images (MRI, Fig. 3) at the level of pelvis and hips showed bilateral atrophy and fatty infiltration of gluteus maximus and the tensor fascia lata on both sides as well as the biceps femoris on the right side. MRI at the level of thighs showed atrophy and fatty infiltration of adductor magnus on the left, long heads of biceps femoris, semimembranosus, proximal semitendinosus, and the vastus intermedius. MRI at the level of calves showed very mild degenerative changes of tibialis anterior, tibialis posterior, and soleus.
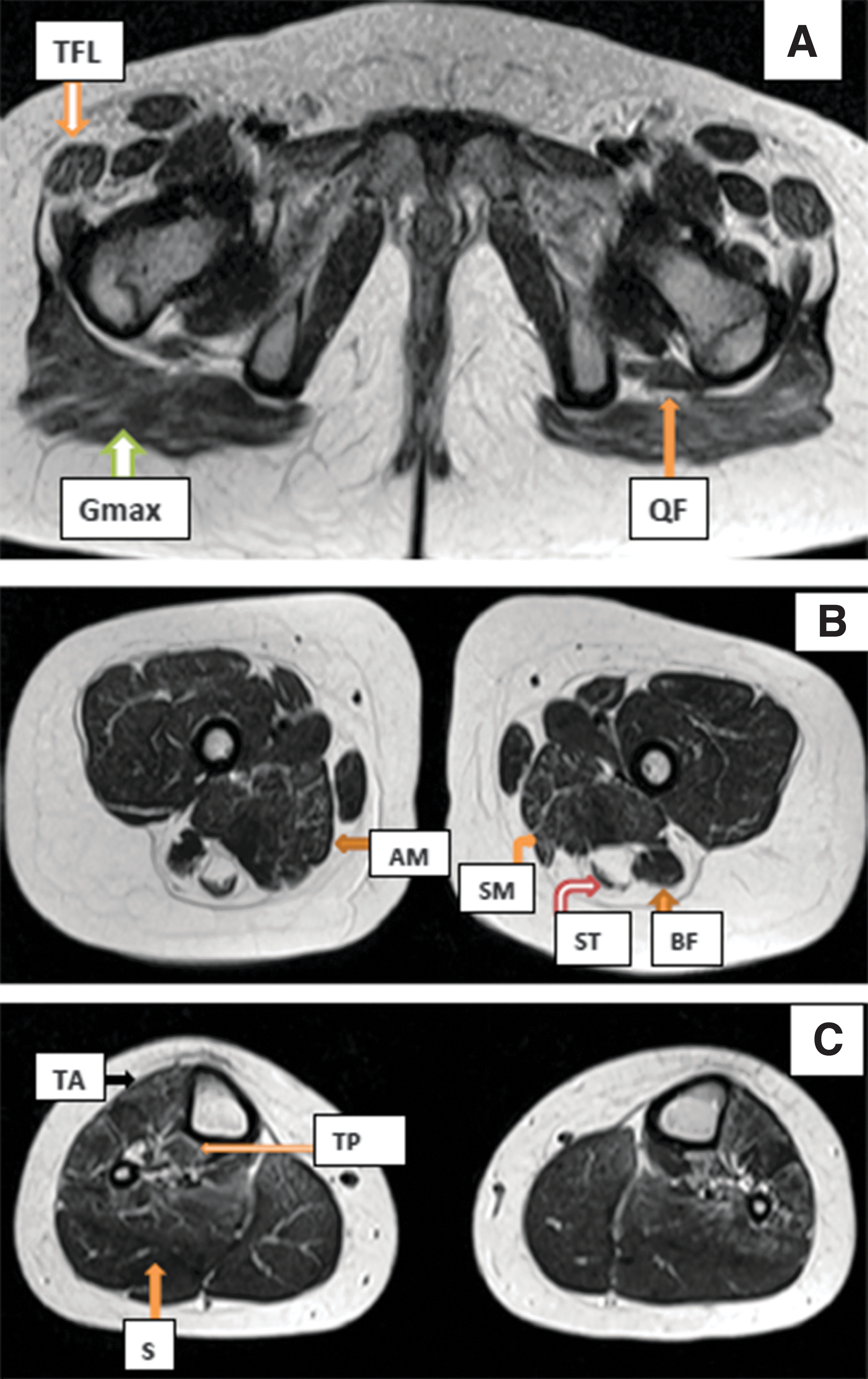
Patient 3 MRI at
Open muscle biopsy, performed at age 7 and taken from the vastus lateralis, showed variation of fiber size with scattered atrophic fibers. Increased internal nuclei were noticeable and estimated to be around 15% of the fibers (Fig. 4A). There was mild increase of endomysial fibrosis and type I fiber predominance. No necrotic or regenerating fibers were seen. The trichrome stain highlighted rare darkly stained areas within the muscle fiber sarcoplasm (Fig. 4B). Oxidative histoenzymatic reactions (nicotineamide adenine dinucleotide-tetrazolium reductase) revealed areas of altered oxidative reactions both devoid of activity within the sarcoplasm of the fibers (Fig. 4C) or showing hyperintensity. This corresponded to the ultrastructural feature of scattered myofibrillar disarray foci with clear small areas of disorganization affecting a few or several sarcomeres with M-line disruption and loss of thick filaments (Fig. 4D).

Pathological evaluation of skeletal muscle of Patient 3.
Sample collection and DNA Isolation
Five milliliters of peripheral blood samples from the consented patients was drawn into EDTA tubes. The Gentra® Puregene DNA Purification Kit was used to isolate genomic DNA from blood samples following the manufacturer's instructions (Gentra Systems, Inc., Minneapolis, MN). The DNA concentration was determined using NanoDrop® ND-1000 (NanoDrop, Inc., Wilmington, DE).
PCR and targeted Sanger-based sequencing
Purified DNA samples were amplified by PCR. First, gene-specific exonic with intronic junction primers were designed using Primer 3 web tool. The primers were optimized and PCR efficiency was determined using normal human DNA as control. After successful PCR, amplified DNA was utilized for sequencing. One of the primers (either forward/reverse) labeled with M13 universal primers was used during the sequencing reaction. Direct sequencing of PCR products was performed on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA) according to the manufacturer's recommendations.
GeneChip Axiom assays, genotyping, and autozygosity mapping
Genome-wide genotyping was undertaken using Affymetrix GeneChip® Human Axiom Array (Thermo Fisher-Affymetrix, Inc., Santa Clara, CA). The assay preparation, chip hybridization, washing, scanning, and primary data analysis were all performed according to the manufacturer's protocols (Affymetrix, Inc.).
Exome sequencing
Exome capture and library construction were done using Agilent Sureselect, and all Exons V5 (50 Mb) Capture Kit (Agilent Technologies, Santa Clara, CA) for library preparation captured libraries were then sequenced on Illumina HiSeq 2500 Sequencer, and mapped against UCSC hg19. The variant detections were performed using freely available tools.
Variants filtering
A comprehensive filtering of the detected variants was done as previously published (AlMuhaizea et al., 2020a, 2020b; Chelban et al., 2020; Seidahmed et al., 2020). Particularly, homozygosity, coding, and splicing, within the autozygome of the index cases in each family were considered. Furthermore, publicly available databases as well as a comprehensive local database for common and rare variants were utilized. The presence (very rare) or absence of the variant in our population was taken into account.
In silico pathogenicity analysis
Two widely used tools, Mutation assessor and Combined Annotation-Dependent Depletion (CADD), were utilized to assess the pathogenicity of the variants.
Results
Genetic findings
Family 1
Genomic DNA samples from affected individuals and parents (Fig. 2A) were run for whole-exome sequencing (WES) and genotyped using genome-wide single nucleotide polymorphism (SNP) markers. For the autozygome detection, AutoSNPa software was used with the software's default settings. The markers pointing loss of heterozygosity were interrogated. All the detected blocks (runs of homozygosity [ROH]) were analyzed during the filtering steps. Among the ROH, a major block was detected on the long arm of chromosome 2 (Supplementary Fig. S4). A homozygous variant was detected in TTN (NM_133378.2:c.C98456G; p.Ser32819Ter) (Fig. 2B, C). The variant is currently NM_001267550 .1(TTN_v001): c.106160C>G:p.Ser35387*.
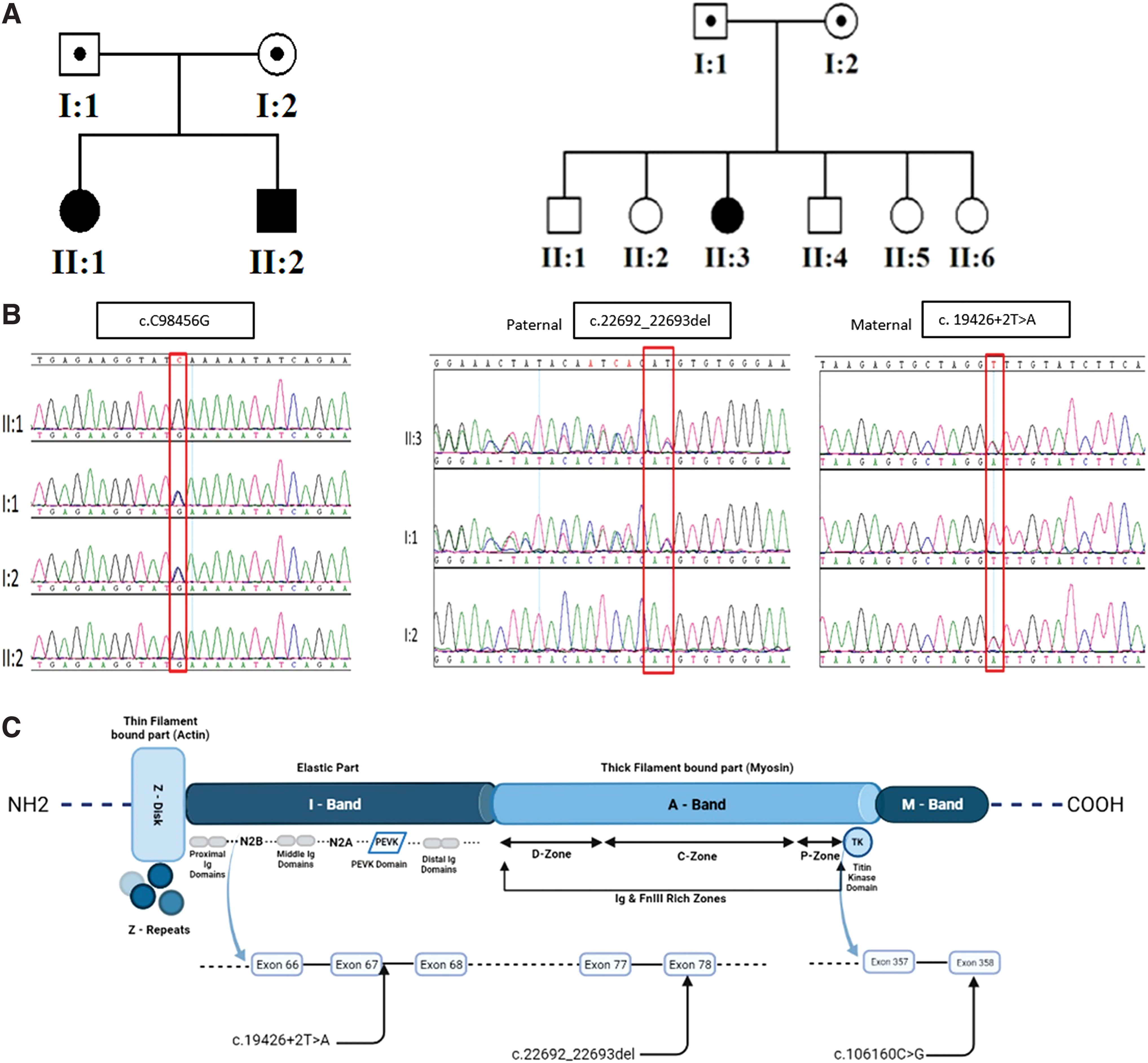
Genetics.
This specific variant has not been previously reported or described and not found in public databases. To further assess the variant's outcome, we utilized two different tools to check the pathogenicity (Reva et al., 2007; Kircher et al., 2014; Schwarz et al., 2014; Laddach et al., 2017; Rentzsch et al., 2019). Mutation Assessor predicted this variant to be a nonsense variant leading to premature stop codon (Supplementary Fig. S5). CADD also predicted variant as being deleterious and disease causing with high score (PHRED:65) (Supplementary Fig. S6).
It is highly probable, according to our current understanding and considering previously reported patients with a similar genotype (Carmignac et al., 2007; Oates et al., 2018) that this variant may still be correctly located in the sarcomere, but of smaller size (basically lacking most of the C-term/M-band portion).
Considering the possibility of founder effect of the variant, we also calculated the predicted age of it according to previously published approaches (Al-Hassnan et al., 2015; AlMuhaizea et al., 2020a; Chelban et al., 2020; Seidahmed et al., 2020). Briefly, bordering distal and proximal SNPs were used for calculation (distal: rs12233083 and proximal: rs6740467). Recombination rates (∼17.7 CentiMorgan) were taken from deCODE Map using SNP positions (GRCh37/hg19 Assembly). The predicted age of the mutation was found to be ∼300 years spanning roughly 12 generations.
Family 2
Two heterozygous variants were detected in Patient 3. These were a previously unreported deletion c.22692_22693del (p.Val7566Glyfs10) in exon 78, which has never been seen before and a splicing variant affecting a canonical splice site (c.19426 + 2T>A in intron 67), which has never been reported. This splicing variant is listed in ClinVar. SpliceAI predicts a very high probability (over 96%, delta score for donor loss = 0.9643) of the variant being splice altering (Jaganathan et al., 2019). Segregation analysis revealed that the parents were heterozygous for one of the two identified variants; the frameshift was on the paternal allele and the splicing variant was on the maternal allele (Fig. 2B).
Discussion
In the present study, we describe the phenotype and highlight the genetic data of three patients with congenital recessive titinopathies. The phenotypic features (Table 1) in Family 1, with two affected siblings, are quite similar to those observed in Salih myopathy (Hackman et al., 1993) first described in consanguineous families of Arab descent originating from Sudan and Morocco (Carmignac et al., 2007). These include muscle weakness (manifesting in early infancy) and delayed motor development and mild facial weakness and relative calf hypertrophy (observed in the older sibling, Patient 1). Other similar features observed in both patients included high arched palate, weak neck flexors, proximal muscle weakness with limb-girdle distribution, moderate joint contractures, and spinal rigidity. No ptosis was seen in either of the siblings.
However, in previously described Salih myopathy patients by Carmignac et al. (2007), ptosis was seen in the majority (4/5), but not all patients (Perrin et al., 2020; Rentzsch et al., 2019). Both patients manifested predominant proximal involvement, whereas of the five patients described by Carmignac et al. (2007), three Moroccan children also showed distal involvement of the tibialis anterior and peroneal muscle with relative preservation of the quadriceps muscle.
Serum CK was mildly raised (about 2.9x normal) in both children, within the range of 1.5-7-fold reported in Salih myopathy (Carmignac et al., 2007). ECG revealed left axis deviation (left anterior fascicular block), signaling cardiac involvement, as has been reported in patients with Salih myopathy as early as age 4 years (Hackman et al., 1993). Dilated cardiomyopathy, signaling severe cardiac dysfunction, manifests between ages 5 and 16 years. However, the patients in Family 1 are 9 and 4 years of age, respectively, which is still below the maximum age limit of manifesting dilated cardiomyopathy (Hackman et al., 1993). A more in-depth investigation of cardiac phenotype, namely cardiac magnetic resonance, was judged to be unjustifiable since it may require general anesthesia.
The phenotype of Patient 3 (Table 1) differs from that described in Salih myopathy since she had normal early motor development. She also revealed no facial weakness or contracture deformity at the age of 9 years, and ECG was normal. Nevertheless, she had proximal muscle weakness with limb-girdle distribution, high arched palate, joint laxity, spinal rigidity, and mild scoliosis. Serum CK was within the normal range, similar to that in the majority of reported patients with congenital titinopathy (Oates et al., 2018). Muscle MRIs (Fig. 3) are similar to the reported findings in congenital titinopathy due to compound heterozygous or homozygous TTN mutations (Yu et al., 2019).
Myopathological and ultrastructural changes in muscle biopsy (Fig. 4) were also similar to those reported in patients with early-onset myopathy due to homozygous or compound heterozygous TTN mutations.
These consisted of central nuclei, areas of altered oxidative reactions devoid of activity, dark oxidative areas, mild endomysial fibrosis, type 1 fiber predominance, and ultrastructural feature of scattered myofibrillar disarray foci (Chauveau et al., 2014; Avila-Polo et al., 2018). WES revealed novel heterozygous variants, including a deletion and splicing variant. This splicing variant affects a canonical splice site probably changing the normal splicing of TTN.
In conclusion, difficulties in analyzing titin protein variants are well recognized and the importance of an integrated diagnostic approach, including muscle imaging and histopathology have been ascertained (Salih, 2012; Chauveau et al., 2014; Salih and Kang, 2020). The novel variant in Patients 1 and 2 (within the first three M-line-encoding exons Mex1, Mex2, and Mex3 of TTN), and the phenotype adds to the other reported cases of Salih myopathy (OMIM 611705). Patient 3 with novel TTN variants in compound heterozygosity depicted the clinical, MRI, and pathologic features of other congenital titinopathies.
Footnotes
Authors' Contributions
M.A.S. and N.K. conceived of the study, and participated in its design and coordination and drafted the article. M.H.H. participated in the collection of the clinical data. M.S., N.K., A.J.A., M.A.S., A.A.B., H.A., and B.U. participated in the interpretation of the genetic results, their relevance, and in the writing of the article. D.C. carried out the age of mutation analysis. N.K., D.C., H.A. carried out in silico analysis for the variants. I.A. studied the radiology. A.S.A. performed the cardiac evaluations. H.A. studied the pathology. All authors read and approved the final article.
Acknowledgments
The authors are grateful to the families and patients for their participation. This research was conducted through intramural funds (RAC# 2120022, 2180004, 2110006) provided by King Faisal Specialist Hospital and Research Center (KFSHRC). The authors thank the KFSHRC Genotyping and Sequencing Core Facilities at Genetics Department, Research Advisory Council Committees, Saudi Human Genome Program and Purchasing Department (Mr. Faisal Al Otaibi) for facilitating and expediting their requests. The authors also thank Dr. Bedri Karakas for carefully editing the article and are greatful and thankful for the generous support that they received from King Salman Center for Disability Research (Project#2180 004 for Dr. Kaya) and King Abdulaziz City for Science and Technology (KACST#14-MED2007-20).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
M.A.S. was supported by the Researchers Supporting Project number (RSP-2020/38), King Saud University, Riyadh, Saudi Arabia. This research was conducted through funds from King Abdulaziz City for Science and Technology (KACST#14-MED2007-20) and King Salman Center for Disability Research (RAC#2180004). The funding bodies have no role in the design of the study, collection, analysis, interpretation of data, and in writing the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.