
Abstract
Objective:
The aim of this study was to determine whether the methylation patterns of the breast cancer-specific gene 1 (BCSG1) and the breast cancer susceptibility gene 1 (BRCA1) can be used as biomarkers for predicting the occurrence and development of breast cancer.
Methods:
Methylation-specific polymerase chain reaction (PCR) was used to detect the methylation status of the BCSG1 and BRCA1 genes in ductal infiltrating carcinomas of the breast; carcinoma in situ of the breast; fibroadenoma of the breast and adjacent normal tissues. Quantitative real-time PCR and immunohistochemistry were used to detect the expression levels of BCSG1 and BRCA1. The BCSG1 and BRCA1 genes were knocked down by siRNA to study their effect of BCSG1 and BRCA1 on the behaviour of breast cancer cell lines.
Results:
The BCSG1 gene was hypomethylated in breast cancer tissues, and its mRNA as well as its protein levels showed elevated expression compared to normal adjacent tissues. In contrast, the BRCA1 gene was hypermethylated in breast cancer tissues and showed correspondingly decreased mRNA and protein expression levels. In vitro experiments demonstrated that BCSG1 could promote the proliferation and migration of breast cancer cells. After inhibiting the methylation, the expression of both the BCSG1 and BRCA1 genes were increased.
Conclusion:
Abnormal methylation patterns of the BCSG1 and BRCA1 genes are associated with the development of breast cancer. Thus, methylatedion analyses of these genes have biomarker potential for breast cancer prognoses.
Introduction
Breast cancer is a malignant tumor occurring in the mammary gland epithelium; it is the most common malignant tumor threatening the physical and mental health of women. In recent decades, the incidence of breast cancer throughout the world has been on the rise. Up to 2020, female breast cancer has surpassed lung cancer to be the world's most common cancer, which is the leading cause of death in most countries worldwide (Sung et al, 2021).
In 2020, there were estimated 2.3 million new cases and 685,000 deaths of breast cancer in women, accounting for 24.5% and 15.5% of the total new cases and deaths of cancer in women, respectively (Heer et al, 2020). Although the incidence and mortality of breast cancer in Chinese women are lower than the world average, the incidence of breast cancer has an upward trend, and the disease burden should not be underestimated (Lei et al, 2021). Thus, the medical research field faces a daunting challenge in exploring the pathogenesis and discovering effective methods for improving the survival rate of breast cancer patients.
The breast cancer-specific gene 1 (BCSG1) is a gene that is highly expressed in advanced breast cancer, and multiple studies have shown that its overexpressed BCSG1 is related to breast cancer progression and metastasis. In addition, BCSG1 can promote the survival of tumor cells in an adverse environment and weaken the effect of chemotherapy (Tang et al, 2016). Studies have shown that the methylation state of the CpG island in the first exon region of the BCSG1 gene is one of the main mechanisms affecting its transcription, which means that the DNA methylation status correlated to the expression of the BCSG1 gene in tumor tissues (Gu et al, 2008).
Moreover, the expression level of the BCSG1 gene is closely related to its methylation state (He et al, 2012, Renz et al, 2018). Therefore, the BCSG gene's methylation status can be used as a marker of breast cancer and is likely one of the drivers of carcinogenesis.
The breast cancer susceptibility gene 1 (BRCA1) is an important breast cancer susceptibility gene and tumor suppressor gene (Gupta et al, 2003). Its encoded protein severs multiple functions to regulate DNA repair, cell division, and chromatin remodeling (Romagnolo et al, 2015, Somasundaram, 2003). BRCA1 germline gene mutations have been associated with familial breast cancer susceptibility, whereas its aberrations are rarely discovered in sporadic breast cancer (Kwong et al, 2016, Van Der Looij et al, 2000).
However, the expression of BRCA1 is significantly decreased in sporadic cancer tissues, indicating a close relationship with its epigenetics. What is more, the molecular pathological features of hypermethylated BRCA1 in breast cancer are still little known.
DNA methylation is an important aspect of epigenetics. The most common DNA methylation is the covalent modification process in which a methyl from adenosine methionine is added to a CpG dinucleotide cytosine at number 5 the carbon atom via the catalysis by methyltransferase (Klutstein et al, 2016, Moore et al, 2012). This form of methylation is conserved quasi-stably during cell division. The methylation patterns of multiple genes are postulated to play an important role in the tumorigenesis of breast cancer (Arechederra et al, 2018, Mahmoud and Ali, 2019, Paço et al, 2020, Pasculli et al, 2018, Rahman et al, 2019).
Materials and Methods
Clinical specimens and cell lines
Breast cancer tissues with complete pathology and clinical data archived in the Biological Sample Center of Anyang Cancer Hospital and Zhengzhou University Affiliated Cancer Hospital that had been collected from January 1, 2006 to December 31, 2018 were included in this study. This collection comprised 30 cases of invasive ductal carcinoma and adjacent tissues. The samples mentioned earlier were quickly maintained with liquid nitrogen for mRNA and methylation level detection. None of the patients underwent treatment before surgery; and none had any family history of breast cancer. The study was under approval from the Institutional Review Board of the First Affiliated Hospital of Zhengzhou University.
Written informed consent was obtained from all patients or their relatives. Pathological diagnoses were made according to the American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 2010 (7th edition) guidelines. The cell lines of MDA-MB-231, BT474, MDA-MB-468, SKBR-3, MCF7, and MCF10A used in this study were obtained from ATCC and cultured in RPMI 1640 (Hyclone) supplemented with 10% fetal bovine serum (Gibco, Australia) at 37°C with 5% CO2.
Extraction of RNA, reverse transcription-polymerase chain reaction
Triazole reagent (Takara) was used to extract RNA from tissues and cells according to the manufacturer's instructions. The total RNA content and purity were determined by a spectrophotometer, with an OD260/OD280 ranging from 1.8 to 2.0. Reverse transcription-polymerase chain reaction (RT-PCR) was performed using the AMV reverse transcription kit from Shanghai Shenggong Biotechnology Technology Services Co., Ltd. according to the manufacturer's instructions.
Oligo (DT18) was employed as a universal primer for reverse transcription. Reagents were immediately added in accordance with the ratio of AMV reverse transcription kit instructions of Shanghai Shenggong Biotechnology Technology Services Co., Ltd., and Oligo (DT18) was reverse-employed as a universal primer for reverse transcription. The products were stored at −20°C. The subsequent PCR amplification of the RT products was performed according to the kit manufacturer's instructions in a volume of 20 μL.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (qRT-PCR) was used to analyze the expression level of the BCSG1 and BRCA1 genes in breast cancer samples. The qRT-PCR analysis was carried out in triplicate by utilizing the SYBR Green I dye (Takara). qRT-PCR was used to analyze the expression level of BCSG1 and BRCA1 in breast cancer samples. The expression levels of BCSG1 and BRCA1 were normalized to GAPDH.
The results are displayed as the difference in tumor tissue expression compared with the expression level in the matched adjacent normal tissue. The Formula Folds = 2−ΔΔCt was used to calculate the relative expression levels of BCSG1 and BRCA1 in tissues. ΔCt values were used to compare expression levels of BCSG1 and BRCA1 in the tumor and control groups. ΔCt = Ct BCSG1 or BRCA1 ± Ct GAPDH, ΔΔCt = ΔCtTumor ± ΔCtNormal. The primers used for qPCR are listed in Table 1.
The Primers Used for qPCR
BCSG1, breast cancer-specific gene 1; BRCA1, breast cancer susceptibility gene 1.
Methylation-specific PCR
Genomic DNA was extracted from cells and tissues using the Wizard Genomic DNA Purification Kit (Promega). One nanogram DNA was denaturated with 0.3 M NaOH at 37°C for 10 min. After adding hydroquinone (Sigma) and sodium bisulfate (Sigma), all samples were incubated at 50°C for 16 h. Methylation-specific PCR (MSP) was performed to analyze the methylation status of CpG-rich regions of the BCSG1 and BRCA1 promoter. The primers used for MSP are shown in Table 2.
Primers Used for Methylation-Specific Polymerase Chain Reaction
The PCR was performed for 30 cycles of 30 s at 95°C, 60°C for 30 s, and then for 10 min at 72°C for a final extension. Finally, PCR products were electrophoresed on 1.5% agarose gel stained with ethidium bromide (Barbieri et al, 2017).
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalin-fixed paraffin-embedded tissue samples. We diluted the BRCA1 (directory: ab238983; Abcam, Cambridge, England) and BCSG1 (directory: ab55424; Abcam) antibodies. The procedure was carried out according to the instructions of SABC-AP (Mouse/Rabbit IgG) kit (Boster Biological Technology). Staining intensity and the degree of staining region were scored using the German semi-quantitative scoring system: staining intensity of nucleus, cytoplasm and/or membrane (no staining = 0; weak staining = 1; moderate staining = 2; strong staining = 3); the range of the percentage of stained cells (0% = 0, 1-24% = 1, 25-49% = 2, 50-74% = 3, and 75-100% = 4). The final immune response score (0-12) was determined by multiplying the two scores.
5-Aza-2′-deoxycytidine treatment
Breast cancer cell lines MCF7 were seeded in culture plates before treatment. Cells were treated in growth medium with 5-aza-2′-deoxycytidine (5-az-DC) (Sigma, St. Louis, MO) at concentrations of 0, 10, 15, and 20 μM, respectively. The growth medium was changed every 24 h for a total of 96 h of treatment (Liu et al, 2016).
Cell proliferation
One thousand cells per well were seeded and cultured in 96-well culture plates with 5 wells per group. Sterile phosphate-buffered saline was added to the hole on the edge of the culture plates to avoid evaporation. CCK-8 reagent (Dojindo Laboratories, Japan) was added to each well and incubated at 37°C for 2h. We then measured the absorbance (OD value) of each well at a wavelength of 450 nm on a microplate reader (Thermo Scientific). We took the average value of each group to draw the proliferation curve; all the experimental results were repeated three times.
Transwell migration and invasion experiments
Two hundred microliters of serum-free cells (0.1-1 × 106/mL) were inoculated into the upper compartment with or without the matrix. One thousand six hundred forty cell culture mediums containing 10% fetal bovine serum were added into each lower compartment. Cells were incubated at 37°C with 5% CO2 for 24 h. Then, the samples were fixed with 4% paraformaldehyde for 20 min, followed by Giemsa staining for 30 min. Next, the upper chamber was wiped with a wet cotton swab and observed by the optical microscope, and five areas were selected for each group.
Western blot assay
Protein was extracted and quantified using the double penicillin acid (BCA) protein quantification kit (KeyGen Biotech). The protein lysate was separated on a 10% SDS-PAGE and transferred to a PVDF membrane (Roche). The membrane was then incubated with specific primary and secondary antibodies. Photos were taken with Pierce ECL Western Blotting Substrate (Thermo Scientific) software. BSCG1, BRCA1, and GAPDH antibodies were purchased from Abclone (Cambridge, MA).
Statistical analyses
We used the SPSS20.0 software (IBM) for statistical analyses. The data are displayed as the mean ± the standard deviation of at least three independent experiments. To look at the differences between two experimental groups, a two-tailed Student's t-test was used. A p-value of <0.05 was considered significant: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
The expression of BCSG1 and BRCA1 in breast cancer
To investigate the role of BCSG1 and BRCA1 in breast cancer tumorigenesis, the expression levels of BCSG1 and BRCA1 were detected in ductal infiltrating carcinoma of breast and adjacent normal tissues by qRT-PCR. BCSG1 mRNA expression was higher in ductal infiltrating carcinoma compared with the adjacent normal. The BRCA1 gene mRNA expression showed the opposite trend (Fig. 1A). We further evaluated the expression levels of BCSG1 in the five breast cancer cell lines MDA-MB-231, BT474, MDA-MB-468, SKBR-3, and MCF7, and normal breast epithelial cells MCF10A.
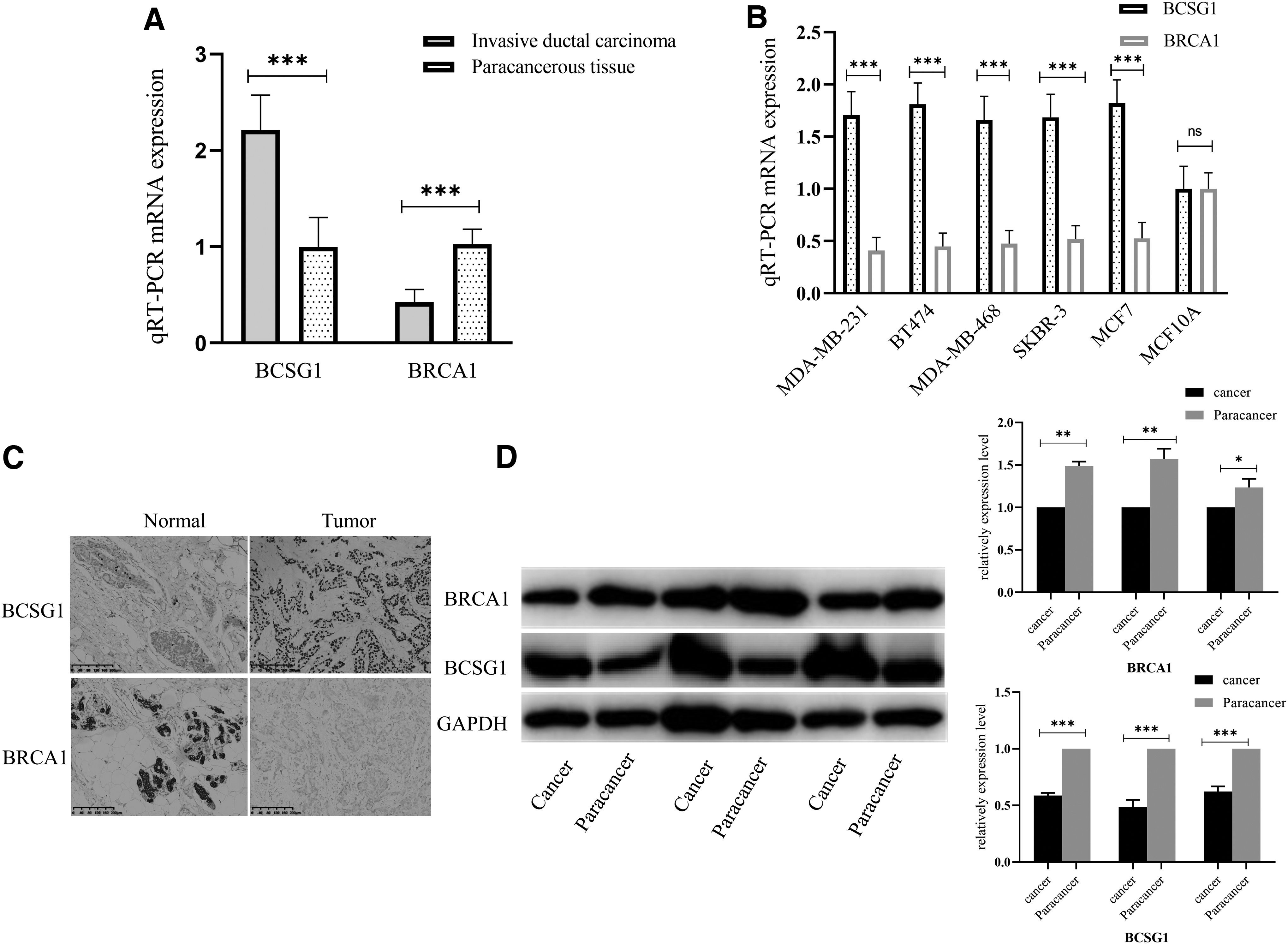
The expression of BCSG1 and BRCA1 in breast cancer.
The expression levels of BCSG1 were also upregulated in the breast cancer cell lines compared with the MCF10A normal breast epithelium cells. The expression of the BRCA1 gene in the cell lines showed the opposite trend (Fig. 1B). Expression of the BCSG1 and BRCA1 protein in cancer and normal tissues was detected by IHC and western blot. These results showed that BCSG1 and BRCA1 protein expression levels were consistent with their cognate mRNA expression levels (Fig. 1C, D).
Methylation levels of the BCSG1 and BRCA1 genes in breast cancer
To explore the methylation status of the BCSG1 and BRCA1 genes in breast cancer, methylation of the BCSG1 and BRCA1 promoter regions was determined by MSP. Compared with MCF10A cells, the methylation levels of BCSG1 were decreased whereas the BRCA1 methylation was increased in all the breast cancer cell lines (Fig. 2A). These results indicate that BRCA1 is silenced by promoter region methylation.
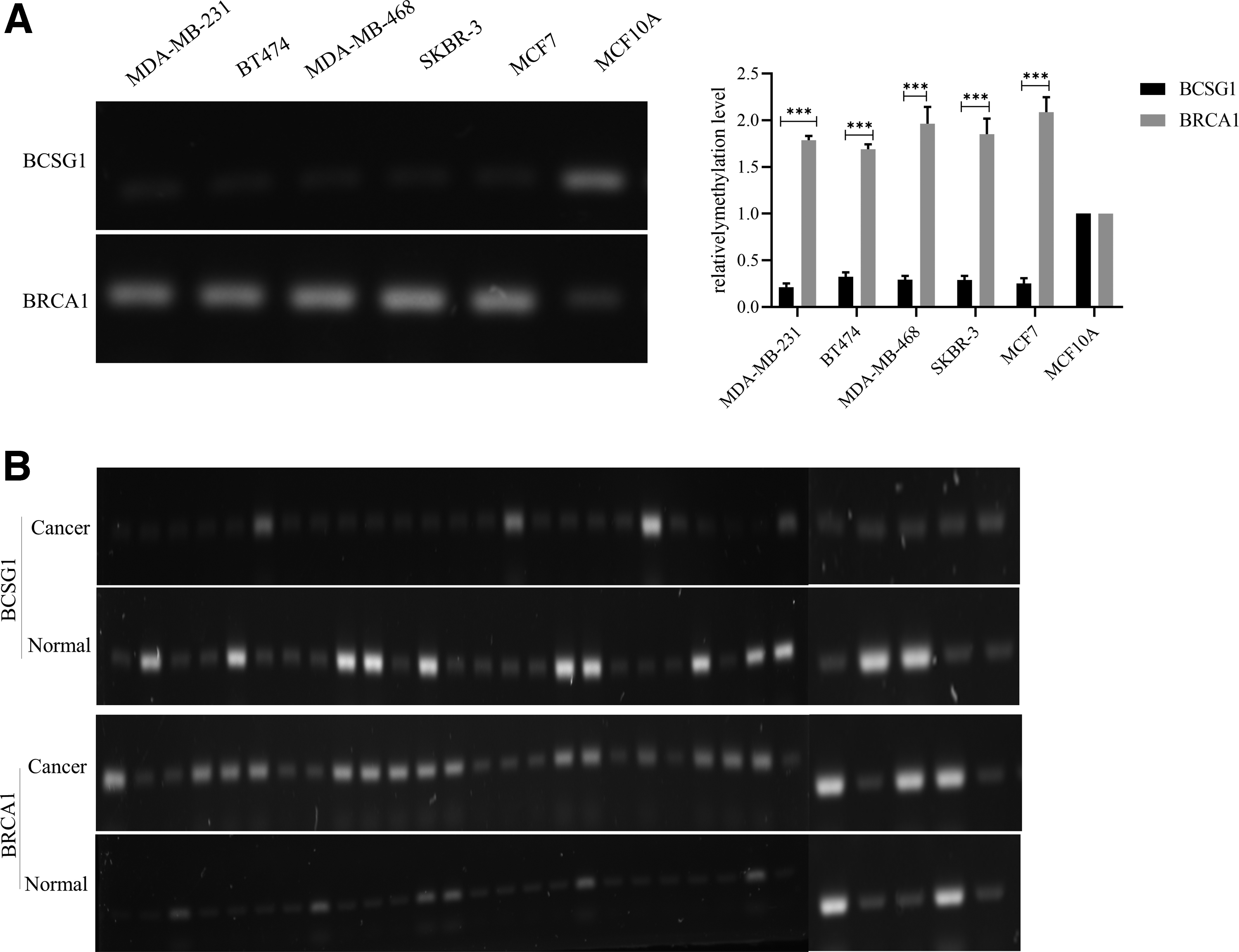
Methylation level of BCSG1 and BRCA1 in breast cancer.
Methylation of the BCSG1 and BRCA1 was also examined in 30 cases of carcinoma of the breast and 30 cases of normal paracancerous breast tissue. The methylation levels of BCSG1 and BRCA1 showed the same patterns in breast cancer tissues as in the cell lines, with the former decreased and the latter increased compared with the adjacent paracancerous tissues (Fig. 2B).
Methylation inhibitors influence the effects of BCSG1 and BRCA1 on the biological behaviors of breast cancer cells
To determine the effect of the methylation status on the function of the BCSG1 and BRCA1 genes in breast cancer, we treated MCF7 cells with the DNA methyltransferase inhibitor 5-az-DC. Our results show that the expression of BRCA1 mRNA increased in a time-dependent manner after the addition of 5-az-DC. There was only a slight increase of BCSG1 mRNA (Fig. 3A). BRCA1 and BCSG1 protein expression also increased after treatment with 5-az-DC in a dose-dependent manner (Fig. 3B).
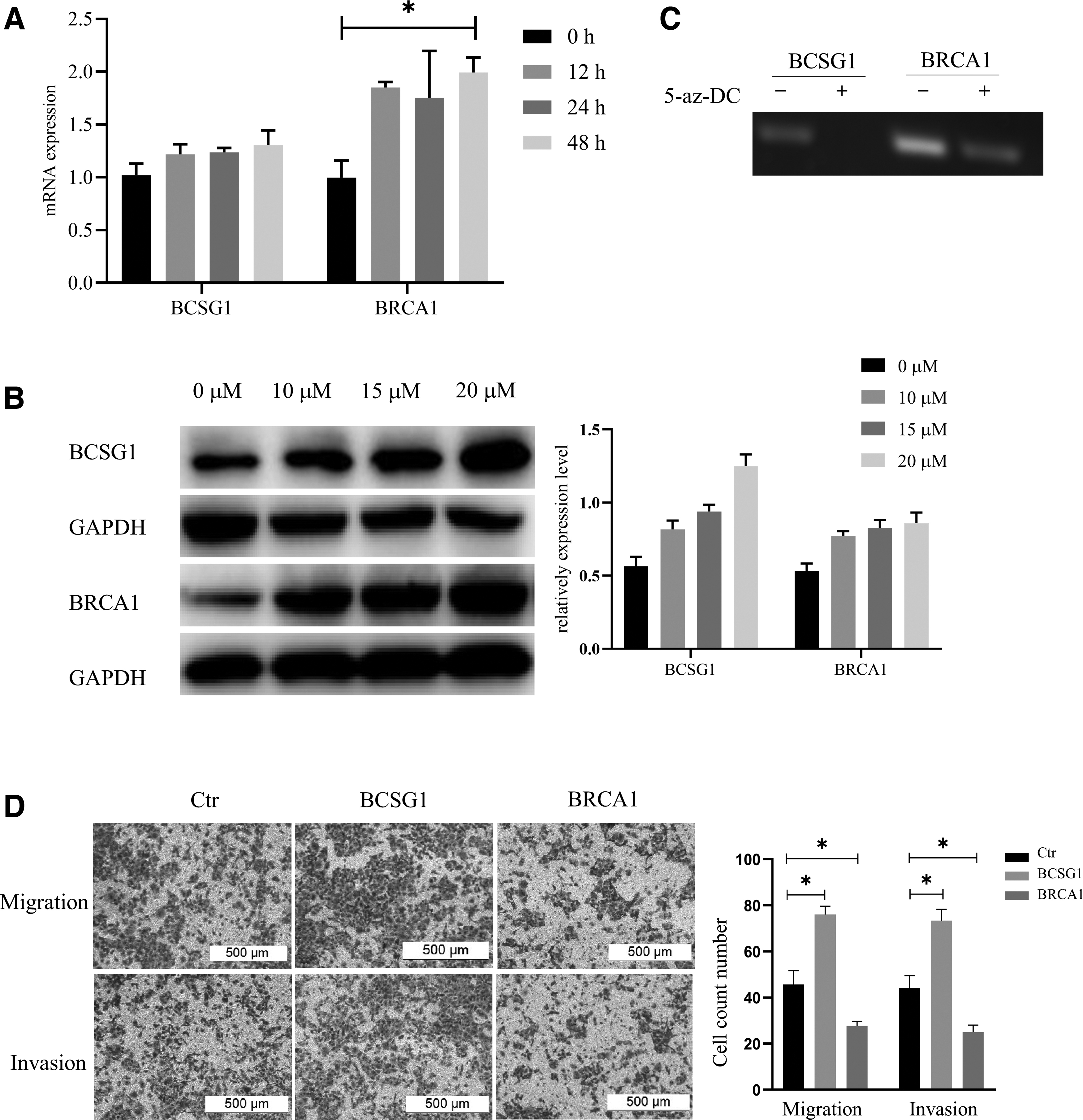
Methylation inhibitors influence the effects of BCSG1 and BRCA1 on the biological behaviors of breast cancer cells.
The methylation state of both the BCSG1 and BRCA1 genes decreased after treatment with 5-az-DC (Fig. 3C). To assess whether the demethylation of BCSG1 and BRCA1 plays a role in breast cancer progression, we analyzed the migration and invasion abilities of MCF7 cells that over-expressed BCSG1 and BRCA1. The DNA demethylation of BCSG1 promotes the migration and invasion of MCF7 cells, whereas the demethylation of BRCA1 has the opposite effect (Fig. 3D).
Effect of BCSG1 and BRCA1 knockdown on the proliferation, migration, and invasion of breast cancer cells
We used si-RNA to construct si-BCSG1-MCF7 and si-BRCA1-MCF7 cell lines to transiently knockdown BCSG1 and BRCA1 expression. mRNA expression of BCSG1 and BRCA1 was significantly reduced compared with the control (Fig. 4A). The proliferation, migration, and invasion ability of si-BCSG1-MCF7 breast cancer cells was significantly reduced. In contrast, the ability of si-BRCA1-MCF7 cells to proliferate and migrate was enhanced (Fig. 4B, C).
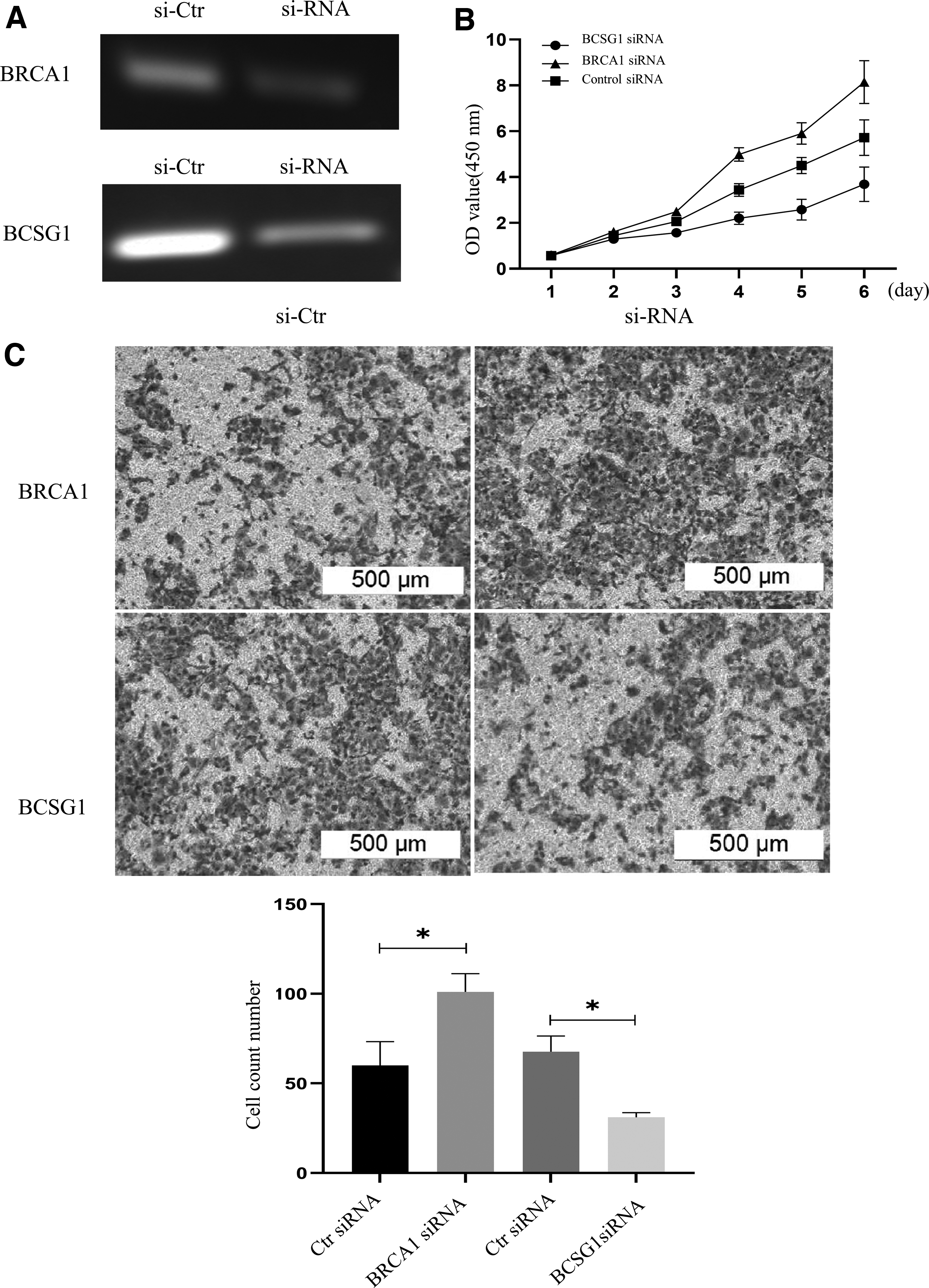
Effect of BCSG1 and BRCA1 knockdown on the proliferation, migration, and invasion of breast cancer cells.
Discussion
BCSG1, also named gamma synuclein (SNCG), has been investigated extensively these decades (Lavedan et al, 1998). The abnormal expression of BSCG1 has been identified in a variety of different tumor types, including hepatocellular, colon, gastric, pancreatic, bladder, and cervical cancers (Liu et al, 2008, Liu et al, 2005, Zhao et al, 2006). Loss of BRCA1 protein expression was widely identified in breast cancer and also related to advanced stage, high grade, and serous disease (Kalachand et al, 2020). In our study, we detected the decreased level of BRCA1 mRNA and protein in both tissue samples and cell lines in breast cancer, whereas the BCSG1 hold the totally opposite trends. This suggests that tumor suppressor gene silencing and abnormal oncogene activation are critical in the malignant progression of breast cancer.
The occurrence and development of breast cancer is a complex process involving multiple genes. Epigenetics refers to modifications to the DNA or histones adjacent to a given gene. These epigenetic changes can alter the expression levels of the affected genes and can be transmitted quasi-stably during the process of replication. One of the principal modes of epigenetic modification involves DNA methylation (Kanwal and Gupta, 2012, Kazemi and Ardekani, 2016, Lee, 2019). DNA methylation takes an S-adenosine methionine as the methyl donor and is catalyzed by DNA methyltransferases DNMTs to bind the 5′-end cytosine of the DNA sequence to methyl groups.
The hypermethylation and hypomethylation of tumor-associated genes is a common occurrence in breast cancer. In this article, the MSP method was employed to detect the methylation status of the BCSG1 and BRCA1 genes in ductal infiltrating carcinoma of breast and adjacent normal tissues tissue and cell lines. As expected, we found that methylation levels were negatively correlated with mRNA expression levels of BCSG1 and BRCA1. The high methylation of BRCA1 and the low methylation of BSCG1 partly explained their expression profiles in breast cancer.
DNA hypermethylation predominately occurs in the CpG islands of DNA promoter regions. Hypermethylation in the promoter region of a variety of tumor suppressor genes is associated with the progression and metastasis of breast cancer (Jandrey et al, 2019, Ng and Yu, 2015). DNA hypermethylation silences the expression of tumor suppressor genes and causes loss of their function to inhibit abnormal cell proliferation and metastasis (Arechederra et al, 2018, Wang et al, 2019). DNA hypomethylation is characterized by a decrease in the overall methylation level of the gene, often resulting in increased expression.
Both in vitro and in vivo experiments showed that BCSG1 could promote the proliferation and migration of breast cancer cells and promote cancer. Inhibition of methylation levels resulted in an increase in BCSG1 expression, but not a significant increase, which may be related to the lower methylation levels of BCSG1 in tumor cells. These results showed that hypomethylation of BCSG1 in tumors is an important cause of its cancer-promoting effect. The methylation status of BRCA1 in breast cancer tissues is opposite to that of BCSG1, and BRCA1 is hypermethylated in tumors leading to its decreased expression. This may be one of the reasons for the loss of its anticancer effect in tumors.
Gene methylation is the third mechanism, in addition to gene mutation and deletion, that leads to the inactivation of tumor suppressor genes. It plays an important role in the occurrence and development of tumors. A change in promoter region methylation patterns is one of the ways that gene expression is regulated at the transcriptional level.
What is more, several studies have uncovered the fact that methylated BRCA1 in breast cancer can be investigated in whole-blood DNA samples with a higher frequency than normal controls (Gupta et al, 2014, Tang et al 2016). The methylated BRCA1 could be a potential biomarker due to the ease of evaluation of whole-blood DNA and the popularization of methylation assay.
In summary, BCSG1 and BRCA1 expression patterns are affected by their levels of DNA methylation in breast cancer tissues and cells. DNA methylation of the BCSG1 and BRCA1 genes correlates with the clinical characteristics of the breast tissues.
Footnotes
Authors' Contributions
G.J. designed the research; J.L. and P.L. performed the experiments and wrote the article; J.L. and H.Y. managed and analyzed the data; G.L. and P.S. collected the samples; and J.L. and P.L. contributed equally to this work.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Funding Information
This work was supported by a grant from Henan Province Medical Science and Technology Research Project (No. 2018021018), Henan Programs for Science and Technology Development (No. 212102310134), and Dr. Pan Li independently received funding from the Youth Innovation Fund of The First Affiliated Hospital of Zhengzhou University, China.