
Abstract
Introduction:
Developmental dysplasia of the hip (DDH) is one of the most common diseases in the pediatric orthopedics, with an incidence of 1-5%. Genetic factors are the bases of the pathogenesis of DDH, but the pathogenic variants and pathogenesis of DDH are still unknown. There are no key accurate diagnostic or prognostic molecular markers for DDH. The purpose of our study was to screen for genetic variant associated with DDH and explore its pathogenesis.
Materials and Methods:
The genetic variation of DDH was tested by variant NGS-based exome analyses, verified by the Sanger sequencing.
Results:
A four-generation family in which DDH was present in three generations was recruited. A novel heterozygous missense variant c.629C>T (p.(Ala210Val)) in exon 7/8 of the parathyroid hormone 1 receptor (PTH1R) gene was identified through screening of two affected and one unaffected family members. The candidate variant was validated in all available family members with all three affected members being positive for the PTH1R variant.
Conclusion:
Our results are highly supportive of PTH1R as a novel candidate gene for DDH and demonstrated that the combination of pedigree information and next-generation sequencing is an effective method for identifying pathogenic variants associated with DDH.
Introduction
Developmental dysplasia of the hip (DDH) is the most common malformation of hip joint in pediatric orthopedics (Feldman et al, 2019; Ponseti, 1978). The incidence of DDH in the neonatal period of North Central Europe (0-28 days) ranges from 1% to 7% (Woodacre et al, 2016). In China, the average incidence of DDH varies from 1% to 10%, but the incidence varies significantly among different races and regions, which is related to genetic factors, environmental impacts, and lifestyle (Pollet et al, 2017).
In the previous study, we found that the prevalence of DDH in Shigatse, Tibet, was ∼174.9/1000 infants (106/606) (Zhao et al, 2019). If not treated early and effectively, children with DDH will suffer from early-onset osteoarthritis (OA) of the hip due to the development of stagnation of femoral head cartilage and acetabulum cartilage and degenerative changes in the early stage, which will easily lead to disability (Tetsunaga et al, 2017). Aronson and his colleague reported that 43% of OA was caused by DDH (Aronson, 1986). One-quarter of total hip replacement under 40 years of age was caused by the delayed diagnosis and treatment of DDH in their infancy (Engesaeter et al, 2011). In view of the high incidence, high disability rate, and good effect of early treatment, the screening, diagnosis, and treatment of DDH have become the focus of pediatric outpatient in all countries (Shaw et al, 2016). However, there is no objective biological indicator for early screening and diagnosis of DDH, and no effective drug to prevent and block the occurrence and development of DDH.
DDH is a complex disorder, and its pathogenesis is still unclear. The etiology of DDH includes both genetic factors and environmental factors (Coleman, 1968; Duman et al, 2019). Environmental risk factors include breech presentation, oligohydramnios, primiparity, and so on (Chan et al, 1997; Stein-Zamir et al, 2008). Epidemiological studies showed that 67.88% of DDH cases were genetically related (Ceylaner et al, 2008). Some genetic studies had shown that DDH was consistent with some characteristics of autosomal dominant inheritance, but it does not show a classical Mendelian inheritance mode (Shi et al, 2012). In the last decade, the advances in the molecular analysis and sequencing techniques allowed researchers to study DDH more thoroughly.
Certain chromosomes, genes, loci, and polymorphisms are being associated with variable severity of this disorder. A recent linkage analysis and an exome sequencing study in a 71-member family pedigree revealed that an rs3732378 variant in CX3 chemokine receptor (CX3CR1) gene on chromosome 3p22.2 was shared by all affected family members and by 15% of sporadic DDH cases, but it was also carried by some unrelated, “married-in” individuals in this family (Feldman et al, 2014, Feldman et al, 2013). Maybe because of its complex etiology, DDH often shows incomplete penetrance (Feldman et al, 2013). So far, no variant in these reported regions has been found to be linked to DDH. Although evidence for the genetic cause of DDH has been widely studied and obtained many results, the exact pathogenesis of DDH remains unclear.
In this study, we identified a novel heterozygous missense variant c.629C>T (p.(Ala210Val)) in exon 7/8 of parathyroid hormone 1 receptor (PTH1R) in DDH patients. This study provided further evidence that DDH is a genetically heterogeneous disease and PTH1R variant accounts for a subset of cases, and provided a direction for developing a sensitive and effective genetic test for DDH.
Materials and Methods
Study oversight and subjects
The protocol and methods of this study were approved by the Institutional Ethical Review Board of the Shanghai Children's Hospital (Shanghai, China, 2018RY001-E01).
A four-generation, 37-member family pedigree from Suzhou city of Anhui province in China of the Han in which DDH was present in three generations and 300 sporadic DDH patients from China was recruited for this study as described earlier (Wang et al, 2018). A pedigree chart was constructed based on the family information

Pedigree of family in this study. Filled-in symbols denote individuals with DDH. Symbols with a backslash denote individuals who died unaffected, male and female. Three members of the family, marked with “exome,” were recruited for exome sequencing analysis. The main proband of the study is indicated by an arrow. DDH, developmental dysplasia of the hip; PTH1R, parathyroid hormone 1 receptor.
Clinical diagnostic criteria
After obtaining written informed consent from all participants or their legal guardians, family members and sporadic DDH patients were diagnosed using detailed clinical examinations and supine anterior posterior radiographs of the pelvis. Results of clinical examinations and radiograph imaging of the hips were evaluated by three pediatric orthopedic surgeons, with clinical opinions of two additional surgeons elicited in any case of disagreement. Radiographic measurements of the hip were taken and affected individuals were identified according to the following criteria: Perkin quadrant (the femoral head is not in the inner lower Perkin quadrant), Acetabular index (>25°), Shenton's line (disrupted), and center edge angle (<20°). None of the subjects assessed had any systemic syndrome. Finally, genetic syndromes involving dysplasia, subluxation or dislocation of the hip, and cardiovascular disease were ruled out and confirmed in this family and in the sporadic DDH patients.
Exome sequencing and criteria for filtering pathogenic variants
Peripheral blood samples from affected/unaffected family members and sporadic DDH patients were collected. Genomic DNA was then extracted from the 23 available family members' and 300 sporadic DDH patients' blood samples using the QIAamp DNA Blood Mini kit (Cat. No. 51104; Qiagen, Hilden, Germany) according to the manufacturer's protocol.
Exome capture was performed using a Sure Select Human All Exon V5+UTRs (Agilent Technologies, New Castle, DE), guided by the manufacturer's protocols. Sequencing was performed using an Illumina Hi Seq 2500 (Illumina, San Diego, CA) following the manufacturer's instructions. A control reference sequence was derived from the 1000 Genomes project and from the reference human genome (GRCh37) assembly of the National Center for Biotechnology Information.
Exome reads were analyzed in a standard bioinformatics pipeline based on Burrows-Wheeler Aligner for sequence alignment on GRCh37 reference, Broad Institute Genome Analysis Tool Kit (GATKv2.6) for genotyping, ANNOVAR 11 and SnpEff for variant annotation, and Exome Depth for copy number variation detection (Cingolani et al, 2012; Li and Durbin, 2010; Plagnol et al, 2012; Wang et al, 2010). Variants were detected using the GATK Unified Genotyper (software.broadinstitute.org/gatk) in conjunction with Southern Han Chinese and Han Chinese in the Beijing exome BAM (compressed binary version of a SAM file that is used to represent aligned sequences) files from the 1000 Genomes Project.
Potentially pathogenic variants were analyzed in two affected individuals (III10 and IV6), and one unaffected individual (II7) (Fig. 1). The screening methods and criteria were performed as described in the reference literature (Adzhubei et al, 2010; Kumar et al, 2009; Schwarz et al, 2010). To refine the list of variants, a filtering strategy was applied with only those variants included that were missense variant, those predicted to be deleterious by SIFT (sift.jcvi.org/), and assumed loss of function variant type (frame-shift deletion/duplication, stop gain, stop loss, and splicing), and those that were not included in the dbSNP137, 1000 Genomes Project, and NHLBI databases.
Sanger sequencing
Candidate variants were validated in the four affected family members and other available family members, as well as in-laws (unrelated individuals married into the family). Exons and adjacent splicing site of the candidate gene were tested using Sanger sequencing in sporadic DDH cases. Polymerase chain reaction products were amplified with specific primers (Table 1) and the primers were synthesized by Shanghai Major bio. Co., Ltd. Reaction mixture (25 μL) contains 2 μL of 200 ng DNA template, 1 μL each of primers (10 μM), and 2 × Premix master mix (Takara, Japan). The PCR was performed with an initial denaturation at 95°C for 1 min, followed by 45 cycles of 94°C for 10 s, 60°C for 20 s, and 72°C for 25 s. The reaction products were analyzed using the ABI 3730XL Genetic Analyzer equipment (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA).
Primers Used to Amplify the Sequences Harboring the Variants in This Study
PTH1R, parathyroid hormone 1 receptor.
Results
Description of the affected family
We have recruited a large four-generation, 37-member family from Suzhou city of Anhui province in China of Han Chinese origin that shows the transmission of DDH through three generations (Fig. 1). Four patients had hip dislocation with all the four radiographic signs and were unequivocally affected by DDH (individual III3, III10, IV2, and IV6). Two affected (individual III10 and IV6) and one unaffected (individual II7) family member were selected for exome sequencing analysis and for analysis of exonic variants and are shown in the pedigree (Fig. 1).
Patient IV6 was diagnosed with DDH by clinical examinations and X-rays radiograph imaging of the hips (Fig. 2). She was the proband of this study and had bilateral hip dislocations. In this family, one affected individual, the mother of IV6, was deemed to be a carrier of variants, and the other affected individual was the grandmother of IV6, who was deemed to be the control. Three individuals had died before this study was initiated (subjects I1, I2, and II3) and one subject died during the study period because of lung cancer (subject II11).
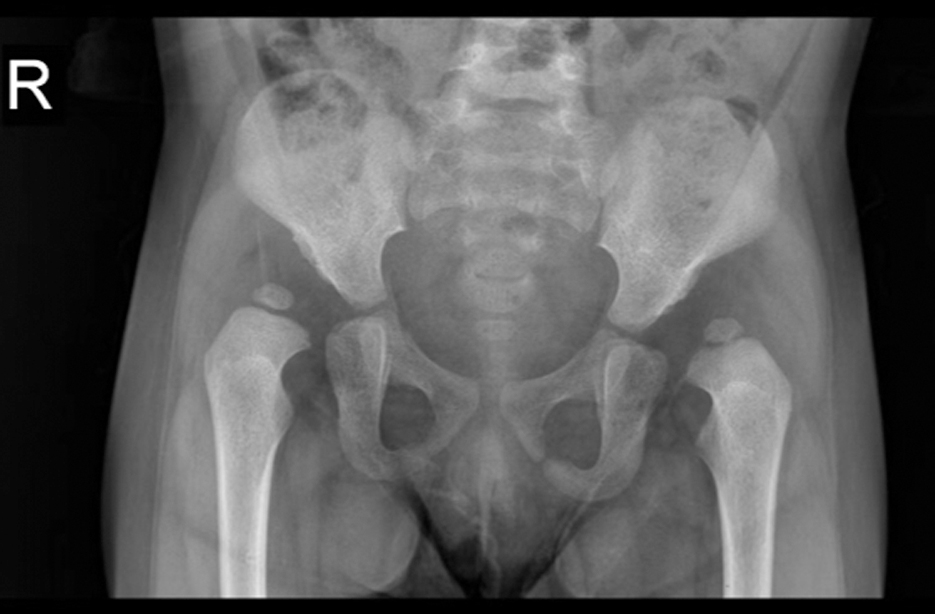
Representative X-ray of the hip joint of DDH patient in this family. Plain radiographs of the hip joint of a proband DDH patient (IV6) in the pedigree, showing bilateral hip dislocations.
Exome sequencing analysis
Exome sequencing analysis of the three family members, including two affected individuals and one unaffected individual in this large family, was performed. An average of 15,974 million 100 bp double-ended sequencing sequence was obtained, with a target area coverage of 98.8% and a sequencing depth of 100 × . A total of 168 heterozygous variants, of which 92.8% were already annotated in a public database (dbSNP v137), were found in two affected individuals and the main proband's grandmother, who was deemed to be the carrier of variants (individuals II7, III10, and IV6).
After three filtering steps, one heterozygous missense variant c.629C>T (p.(Ala210Val)) in PTH1R located on of chromosome 3 has remained finally. The PTH1R variant is the possible candidate pathogenic variant for DDH in this family

The pathogenic gene of DDH was obtained by exome sequencing. Exome sequencing analysis of the five family members, including three affected individuals and two unaffected individuals in this large family, was performed. The variant in PTH1R gene located on chromosome three may be a DDH pathogenic gene.
Potentially deleterious variant assessment and verification using Sanger sequencing
This heterozygous missense variant of PTH1R leads to the change of Alanine to Valine. Although this kind of variant may have no effect on the expression products or may bring benefits, most of them are harmful or even fatal. Potentially deleterious variants were evaluated by its SIFT score. The SIFT value of the PTH1R variant was 0.01, which indicated that the variant site was deleterious and might be damaging to its protein function according to the ACMG/AMP classification (Harrison et al, 2019; Richards et al, 2015). Thus, PP3 could be applied. PM2 that could be used as this variant is absent from population databases (e.g., 1000 Genomes Project, National Heart, Lung, Exome Aggregation Consortium). Three cases were positive for this variant (PP1). Located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation (PM1). So according to ACMG/AMP variant pathogenicity guideline, PTH1R variant has two moderate pieces of evidence for pathogenicity and two support evidence for pathogenicity (2PM +2PP); this variant should classify as likely pathogenic.
Then, candidate variants were confirmed by Sanger sequencing using all available DNA samples from family members. We found that PTH1R variant existed in some DDH patients and unaffected people of this selected family by Sanger sequencing
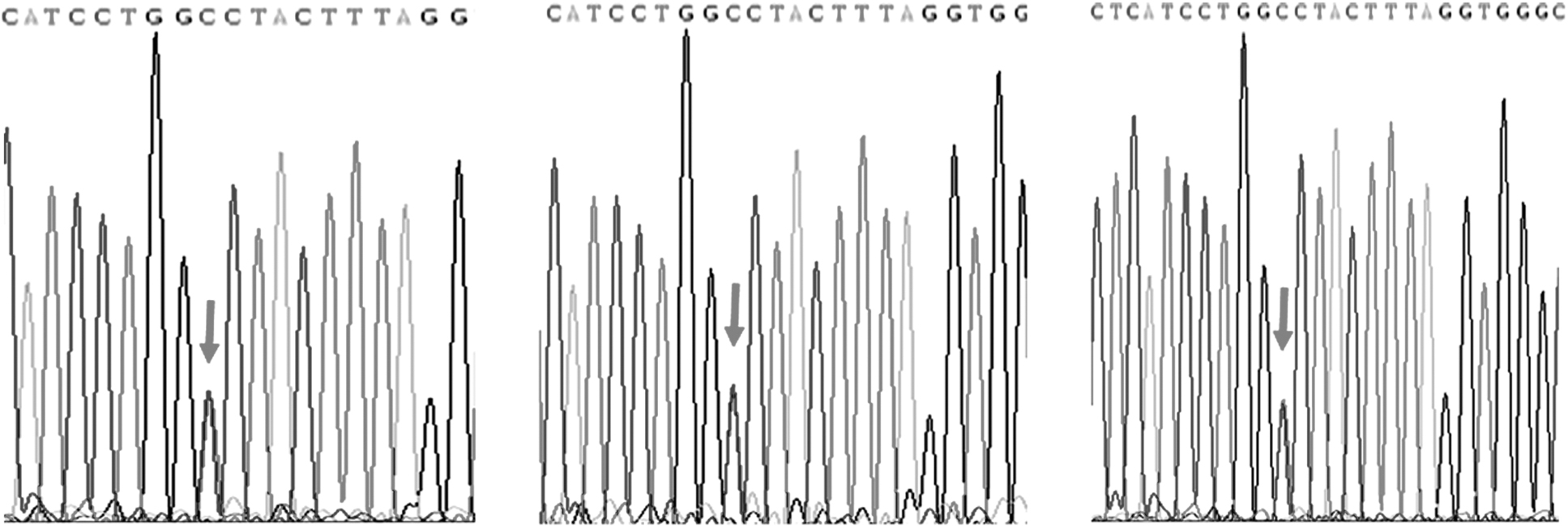
Variant verification in family members with PTH1R variants by Sanger sequencing. Three DDH patients were positive for the PTH1R variant in the pedigree as determined by Sanger sequencing.
Discussion
DDH is the most common malformation of the hip joint in pediatric orthopedics, which is characterized by incomplete formation of the acetabulum leading to subluxation or dislocation of the hip joint. However, the genetic architecture of DDH is poorly understood. There is no key, typical morphological characteristic, and accurate diagnostic or prognostic molecular marker for DDH patients.
DDH has a complex etiology with environmental and genetic causes, and appears to be inherited in an autosomal dominant manner and exhibits incomplete penetrance (Harcke, 1999; Hatzikotoulas et al, 2018).With the application of family linkage analysis and sequencing analysis in DDH, the genetic basis of DDH has been confirmed by more and more studies. The exome sequencing analysis of a Japanese DDH pedigree showed that the pathogenesis of DDH was related to chromosome 13q22, but no pathogenic variant was found in locating genes as PCDH9, DACH, and CDH1, which related to bone and joint development on this chromosome (Mabuchi et al, 2006).
Combined with linkage analysis and exome sequencing analysis, researchers found a variant in CX3CR1 gene located in the 26Mb candidate region of chromosome 3p38.7-41.31 (Feldman et al, 2014). Using the largest DDH genome-wide association study to date, researchers found the heritable component of DDH attributable to common genetic variants to be 55% and distributed equally across the autosomal and X-chromosomes and identified replicating evidence for the association between GDF5 promoter variation and DDH (Hatzikotoulas et al, 2018). Much, however, remains uncertain and challenging. Although DDH is heritable, its genetic architecture remains vague and poorly characterized; up to date, research results of the Chinese population have low repeatability and inconsistent positioning of DDH-related chromosomal abnormalities.
In this study, using high-throughput exome sequencing, we have found a novel variant in exon 7/8 of PTH1R in DDH and it was found to be presented in two affected individuals and one unaffected individual. In addition, candidate variants were confirmed by Sanger sequencing. Results showed that three cases were positive for the PTH1R variants in this family. A variant in CX3CR1 gene was identified by Feldman and his colleagues. However, this variant was also presented in the unrelated normal person spouse of the family through the family verification (Feldman et al, 2013). They also found the ablation of CX3CR1 in the murine model for DDH affected acetabular morphology and gait (Feldman et al, 2017). So our results demonstrated that a novel heterozygous missense variant c.629C>T (p.(Ala210Val)) of PTH1R in preliminary sequencing studies of the 37 large pedigree was closely related to DDH pathogenesis.
Many literatures showed that PTH1R plays an irreplaceable role in the process of bone formation. PTH1R was mainly expressed in bone and kidney, especially in osteoblasts and renal tubular cells, and participates in the mineral ion homeostasis regulation of parathyroid hormone (PTH) or parathyroid hormone-related protein (PTHrP) in these cells (Lavi-Moshayoff et al, 2010). PTH1R, a prominent feature of osteoblast-like cells, plays an important role in the growth and proliferation of osteoblasts and the regulation of calcium and phosphorus metabolism (O'Brien et al, 2008). PTH1R is a specific receptor for PTH and PTHrP, and is a classic B-type G protein-coupled receptor (GPCRs), which can be activated only by binding with PTH or PTHrP and be activated to play different signal transduction functions (Datta and Abou-Samra, 2009; Gardella and Juppner, 2001). In recent years, PTH1R has attracted more and more attention in regulating the proliferation and differentiation of embryonic and postpartum chondrocytes by binding with PTH/PTHrP and activating downstream complex signaling pathways (Harrington et al, 2004).
Jobert et al found that the first 11 amino acids of the fifth transmembrane subunit of PTH1R receptor in children with Blomstrand chondrodysplasia were missing, which made the receptor unable to bind with PTH or PTHrP normally, lose the regulatory role of PTHrP, and accelerate the differentiation of chondrocytes and the process of endochondral osteogenesis (Jobert et al, 1998). In addition, PTH1R knockout mice showed similar growth plate abnormalities, indicating that PTHrP/PTH1R pathway plays an important regulatory role in endochondral osteogenesis (Lanske et al, 1996). PTH1R allele homozygous inactivated mice had severe skeletal dysplasia of limbs, limbs were significantly shorter and thicker (34). Histological observation showed that the growth plate of PTH1R allele homozygous inactivated mice was significantly atrophied, while hypertrophic chondrocytes were significantly proliferated (Lanske et al, 1999). DDH is caused by the abnormal development of bone, cartilage, and soft tissue around the hip joint before and after delivery. Thus, the above results and studies fully indicated that the pathogenic variant of PTH1R is a novel candidate gene for DDH.
There are still some deficiencies in this study as follows: (1) to ensure the more rigor of the study, we should increase the sample size of sequencing and validation. (2) Some functional analysis of PTH1R should be conducted in model cells (like ATDC5 cell) and mice (like knock-in mutant murine).
Conclusion
Our study proposed a novel heterozygous missense variant of PTH1R, which is closely related to the pathogenesis of DDH in a Han Chinese pedigree and in sporadic DDH cases. PTH1R is a novel candidate pathogenic gene for DDH, and this study provided a possible mechanism suggesting that novel PTH1R variants are implicated in the pathogenesis of DDH.
Footnotes
Authors' Contributions
L.H.Z.: design, co-ordinate, and conduct the study. L.H.Z., H.Y., S.Q.W., Q.J., and L.Y.F.: provision of study materials. L.H.Z., D.Y., Z.W.Z., S.Q.W., Q.C.M., J.W., M.J.C., and Y.C.W.: collection and/or assembly of data, conduct the study, data analysis, and data interpretation. D.Y.: article writing.
Availability of Data and Material
The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was approved by the Institutional Ethical Review Board of the Shanghai Children's Hospital (2018RY001-E01). Written informed consent was obtained from all patients and controls who participated in the experiments and from their legal guardians.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was sponsored by Natural Science Foundation of Shanghai (No. 20ZR1446600), Youth Project of Medical Engineering cross Research Foundation of Shanghai Jiao Tong University (No. YG2021QN115), National Major Science and Technology Infrastructure Project of Translational Medicine of Shanghai Jiao Tong University (No. TMSK-2020-123), and Medicine, Multi-center Clinical Research Project of Shanghai Jiao Tong University (No. DLY201825).