
Abstract
Background:
The LIPA gene encodes for lysosomal acid lipase (LAL), which catalyzes the hydrolysis of cholesterol esters and triglycerides. Variations in the LIPA gene impair LAL activity, predisposing patients to a rare metabolic disorder called LAL deficiency (LAL-D). The lack of functioning LAL promotes lipid accumulation and subsequent dyslipidemia, which can increase the likelihood of complications in both infants and adults. Although the worldwide prevalence is 1:500,000 births, the frequency in Mizrahi Jewish populations is projected to be as high as 1 in every 4200 births (Valles-Ayoub et al.) based on the LIPA p.G87V variant frequency among 162 individuals.
Materials and Methods:
This study was conducted to validate the previously reported prevalence of LAL-D in the Mizrahi Jewish population based on the pathogenic LIPA missense variants in exon 4 (c.260G>T; p.G87V) and exon 8 (c.894G>A; p.Gln298=) using a larger cohort of those with Middle Eastern ancestry living around Los Angeles. Among the 1184 individual samples sequenced, 660 self-reported as Mizrahi Jewish, while the remaining 524 came from other Middle Eastern groups labeled as “non-Jewish.”
Results:
Of the 1184 samples, 22 alleles of the exon 4 variant were identified (1.85%), and 2 alleles of the exon 8 variant were identified (0.16%). For the exon 4 variant, 20 of 22 (90.9%) heterozygotes were Mizrahi Jewish, while 2 of 22 (9.09%) heterozygotes were “non-Jewish.” For the exon 8 variant, 2 of 2 (100%) heterozygotes were Mizrahi Jewish. This suggests that the prevalence of LAL-D in this population is 1 in 900, which suggests that LAL-D may be 4.6% higher in the Mizrahi Jewish population in previous reports.
Conclusion:
These findings show increased prevalence of LIPA gene exon 4 variation p.G87V in the Middle East population when compared to the general population, indicating the need for prenatal screening in those of Mizrahi Jewish ancestry.
Introduction
Lysosomal acid lipase (LAL), an enzyme that primarily functions in the lysosome, catalyzes the hydrolysis of cholesteryl esters (CEs) and triglycerides (TAGs). Since LAL regulates lipid and cholesterol balance by binding to low-density lipoproteins (LDLs) and freeing CEs and TAGs for degradation, it is an essential molecular component in lipid metabolism. LAL is encoded by the gene Lipase A (LIPA), located on chromosome 10 (10q23.3) (LIPA lipase A, lysosomal acidtype, 2023). Variations in the LIPA gene can result in LAL deficiency (LAL-D), a metabolism disorder that is inherited in an autosomal recessive manner. Variation in the LIPA gene disrupts CE and TAG intralysosomal degradation, causing an accumulation of lipids in cells and tissues throughout the body (Pastores and Hughes, 2020; Li and Zhang, 2019; Pericleous et al., 2017).
Patients with CE and TAG accumulation present clinically with pathologies in the liver, systemic vasculature, and reticuloendothelial systems. Patients with LAL-D caused by LIPA variations demonstrate distinct clinical presentations such as hepatic dysfunction and dyslipidemia. Specifically, hepatomegaly associated with liver dysfunction and elevated LDL levels is known to be the principal clinical features of LAL-D (Li and Zhang, 2019). Currently, LIPA has several known pathogenic variants that progress to either partial or complete loss-of-function of the enzyme (Fasano, 2012). Diseases caused by LAL-D vary in severity, which is attributable to the type of LIPA variation and its impact on enzyme function. Historically, the rapidly progressive infantile onset variant was termed Wolman's Disease (WD), while the adult-onset variant was termed cholesteryl ester storage disease (CESD).
Due to the lack of properly functioning LAL protein, harmful amounts of lipids accumulate in the spleen, liver, bone marrow, intestine, adrenal glands, and lymph nodes. Neonates with WD are healthy and active, but rapidly develop symptoms of severe malnutrition in the first few months of life, and often succumb to the disease before the age of 1 (Hoffman et al., 2015). Early detection of WD is crucial for the appropriate management and consists of enzyme replacement therapy, bone marrow transplant, and gene therapy (Potter, 2021; Li and Zhang, 2019). Conversely, CESD patients become symptomatic later in life and typically present as adolescents or adults. CESD is also represented by an abnormal lipid profile, liver function abnormalities, or gallstones.
Similarly, early detection is critical for managing CESD and studies demonstrate how a delay in the initiation of therapy could lead to detrimental outcomes such as severe hepatic dysfunction and ultimately cirrhosis (Gómez-Nájera et al., 2015). As such, early detection of LAL-Ds and diagnosis of the disease is critical. Due to the uncommon nature of the disease and lack of significant studies, prevalence rates are not accurate. However, studies have estimated worldwide incidence to be 1/350,000 newborns (U.S. Natioanal Library of Medicine, 2017). Since Los Angeles is known to be a highly concentrated region of expatriates from Iran and other Middle Eastern countries, previous studies focused on the frequency of WD within this community. These studies indicate that 1/4200 newborns of Iranian-Jewish couples may be at risk to develop WD (Valles-Ayoub et al., 2011). Additional studies are required to confirm and further validate the higher frequencies seen in this sample pool and to determine if people of Iranian-Jewish descent and even possibly solely Middle Eastern descent, are at a higher risk for WD.
LAL-D is an autosomal recessive trait; therefore, two copies of the variant, mutated allele are required for active disease. The clinical profile of the patients included in this study did not exhibit the two known diseases that result from these pathogenic variations but are at high risk of being carriers and passing down the variation to offspring.
To better report the prevalence of known missense variations in the LIPA gene across a cohort of Middle Eastern residents of Los Angeles, we analyzed 1184 samples of individuals with Middle Eastern background within the Los Angeles community for the following pathogenic LIPA variations: LIPA gene exon 4 variant c.260G>T and the exon 8 splice site variant c.894G>A which results in the skipping of exon 8 (Aslanidis, 1996; Hoffman et al,, 2015).
Materials and Methods
In our study, we analyzed two variations from 1184 samples of individuals with Middle Eastern backgrounds within the Los Angeles community. The 1184 samples were collected from various Middle Eastern communities in Los Angeles with cultural backgrounds. The sample population comprised individuals of varying Middle Eastern heritages such as Mizrahi Jewish and many other Middle Eastern ethnic groups. The ethnic groups that were not Mizrahi Jewish were classified under “non-Jewish” to have a more even distribution.
The molecular assays defined were performed by Firma Laboratory INC specialized in performing high-complexity clinical molecular genetic testing, regulated under the Clinical Laboratory Improvement Amendments of 1988, California Laboratory Field Services, and accredited by the College of American Pathologists.
Buccal epithelial cells were collected with the Hydra Flock 6″ Sterile Elongated Flock Swab w/Plastic Handle & Dry Transport Tube (Puritan Medical Products, Glendora) cellular DNA was isolated using the Quick-DNA Kit (Zymo Research, Irvine ) following manufacturer's instructions. DNA optical density was measured using the NanoDrop One Spectrophotometer (Thermo Fisher Scientific, Waltham) for concentration determination. After DNA quantitation, DNA was diluted using RNAse-free H2O to a concentration of 20 ng/μL. Isolated DNA was stored at −20°C as needed. Polymerase chain reaction (PCR) amplification was done on LIPA gene exons using BioRad CFX 96 Real-Time System. PCR Primers were designed for the amplification of exon 4 and 8 coding regions of the LIPA gene (Table 1).
Primers Designed for Amplification of the LIPA Gene Exon 4 and 8
The amplification assay included a 3 min enzyme activation step at 94°C followed by 35 PCR cycles, each of which involved three steps: denaturation of DNA template and primers for 30 s at 94°C, annealing for 30 s at 60°C, and extension for 1 min at 72°C. The amplicon was electrophoresed for 30 min at 90 V and viewed in a 4% Nusieve® 3:1 plus agarose to confirm that amplification. The gel was electrophoresed with a 3 μL Genemate Quanti-Marker 1 kb and 3 μL amplicon. The Electrophoresis gel signal was detected on an Epi Chemi II Darkroom (UVP) and viewed using LabWorks (Fig. 1a, b). After amplification was confirmed, the amplicons were then cleaned and concentrated using a ZR DNA Clean & Concentrator™ (Zymo Research, Irvine) kit according to the manufacturer's instructions and eluted with triple-distilled water to a final volume of 30 μL. Optical densities and DNA quantification using the Thermo Scientific NanoDrop One Spectrophotometer.
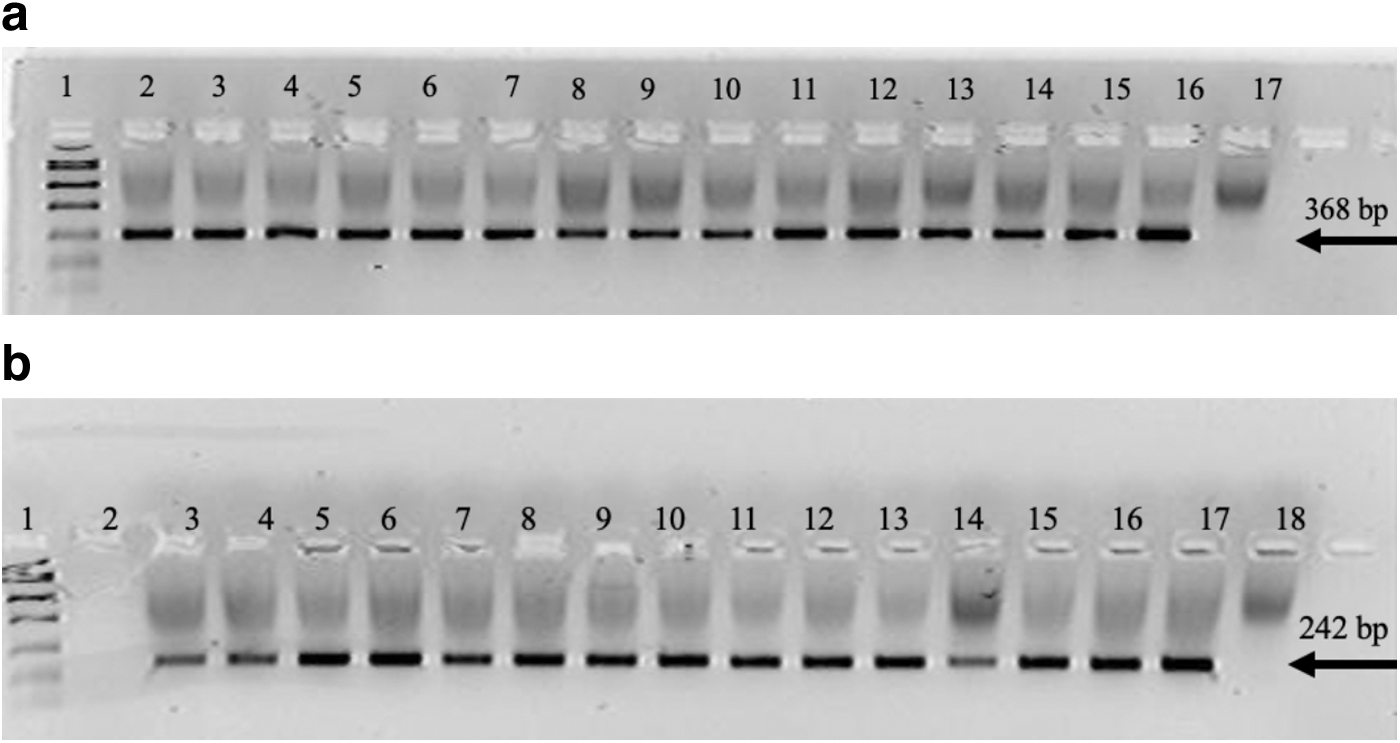
Gene LIPA exons 4 and 8 were sequenced using the Sanger method. A 15 μL reaction mix was prepared for cycle sequencing, comprised of 5 μL of 5 × sequencing buffer, 2 μL of forward and reverse universal primers (1.0 μM), 1 μL of Big Dye Terminator® (Thermo Fisher, West Hills,), and 7 μL of 5 ng/μL amplicon product. Cycle sequencing was attained with an enzyme activation step of 1 min at 96°C followed by 25 PCR cycles, each included denaturation for 1 min at 96°C, annealing for 5 s at 50°C, and extension for 1.5 min at 60°C. Cleaning of the mix using the ZR DNA Sequencing Clean Up Kit™ (Zymo Research) and eluted with 10 μL of HiDi™ Formamide. Eluted samples were diluted 1:10, with a total volume of 10 μL, and loaded for sanger sequencing on the AB (Applied Biosystems) 3130xl Genetic Analyzer. Electropherograms were visually examined for peak shape and height to confirm nucleotide identity using the Sequencher 4.9 software. Results were aligned and compared to reference assembly of human DNA by the Human Genome Sequencing Number (NM_001127605).
Results
Variations of the LIPA gene located on chromosome 10 (10q23.3) can be found across all populations but are prevalent for people of Middle Eastern descent (Valles-Ayoub et al., 2011). This study specifically focused on the pathogenic variants c.260G>T and c.894G>A within the Middle Eastern population of Los Angeles.
Variant c.260G>T was found on exon 4 and resulted in a glycine-to-valine amino acid change (Fig. 4). Variant c.894G>A was found on exon 8 and resulted in a splice junction variant causing the loss of mRNA amino acids (Fig. 5) (Valles-Ayoub et al., 2011). To evaluate the prevalence of the variants c.260G>T and c.894G>A, 1184 samples collected from the Middle Eastern population in the Los Angeles area were analyzed.
Mizrahi Jews made up 55.74% (N = 660) of the study population, while 44.26% (n = 524) consists of other religions that are classified as “non-Jewish” (Fig. 2). Sequencing results showed 24 out of 1184 heterozygotes (2.03%), conferring a carrier frequency of 1 in 49.26 in this population. This correlates with the already predicted estimate of heterozygotes within this population group (Cappuccio et al., 2019). Out of 24 heterozygotes 22 were Mizrahi Jewish (91.6%) and two were “non-Jewish” (8.4%) (Fig. 3). The prevalence of heterozygous exon 4 variation, p.G87V (ggc>gtc
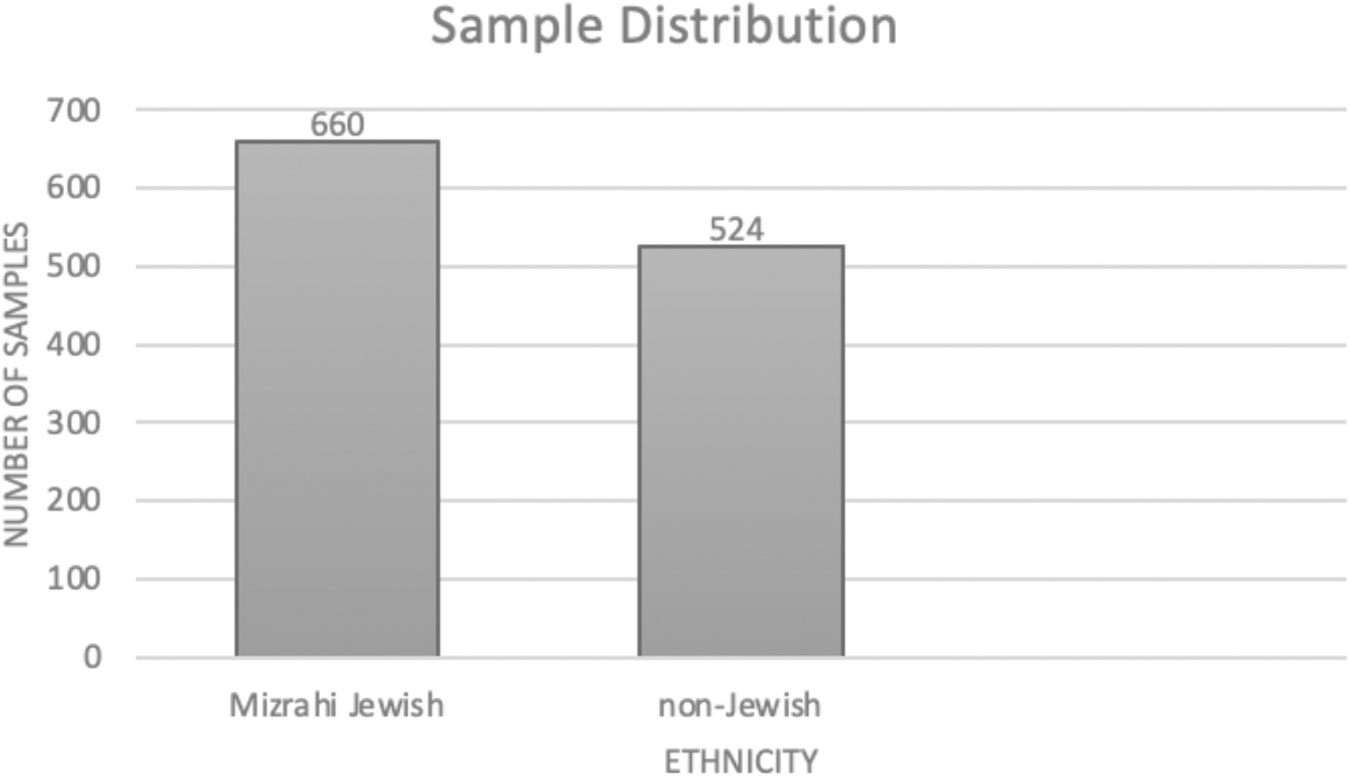
Ethnic distribution for the samples within this study.

Heterozygotes that were found within the study were divided by ethnicity.
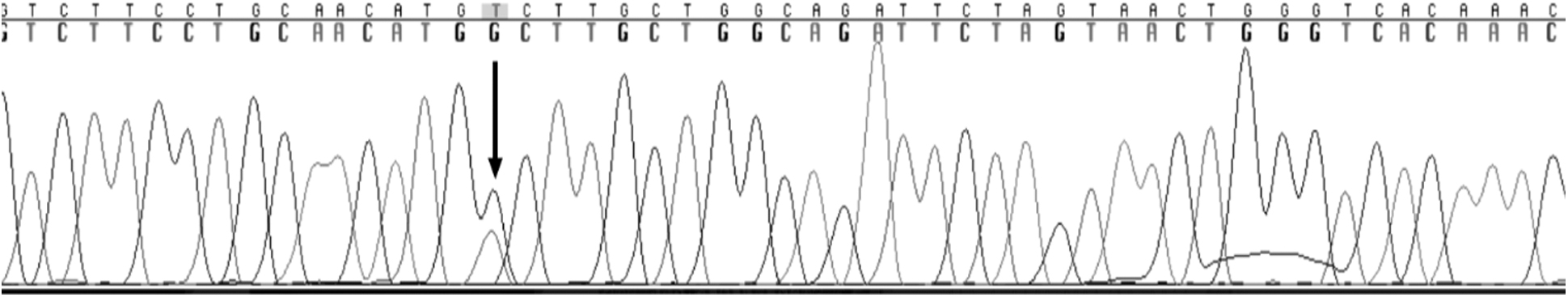
Electropherogram of exon 4 depicting the heterozygous exonic variation (p.G87V ggc>gtc). The variation is marked by the arrow.
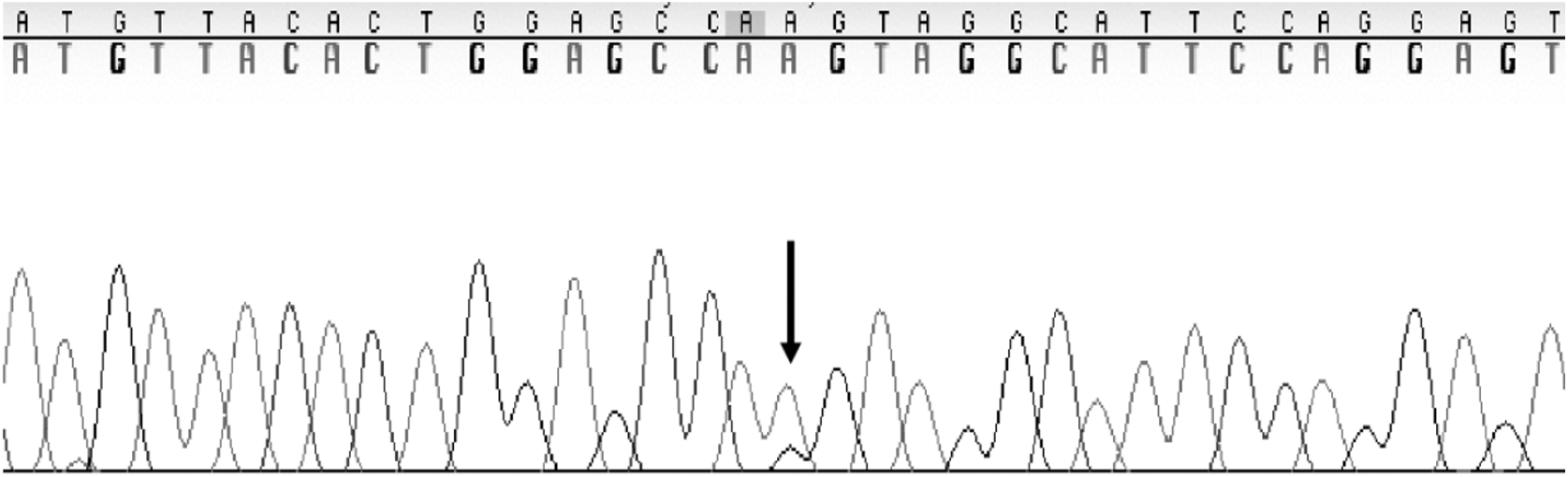
Electropherogram of exon 8 depicting the heterozygous exonic variation (p.Gln298=cag>caa). The variation is marked by the arrow.
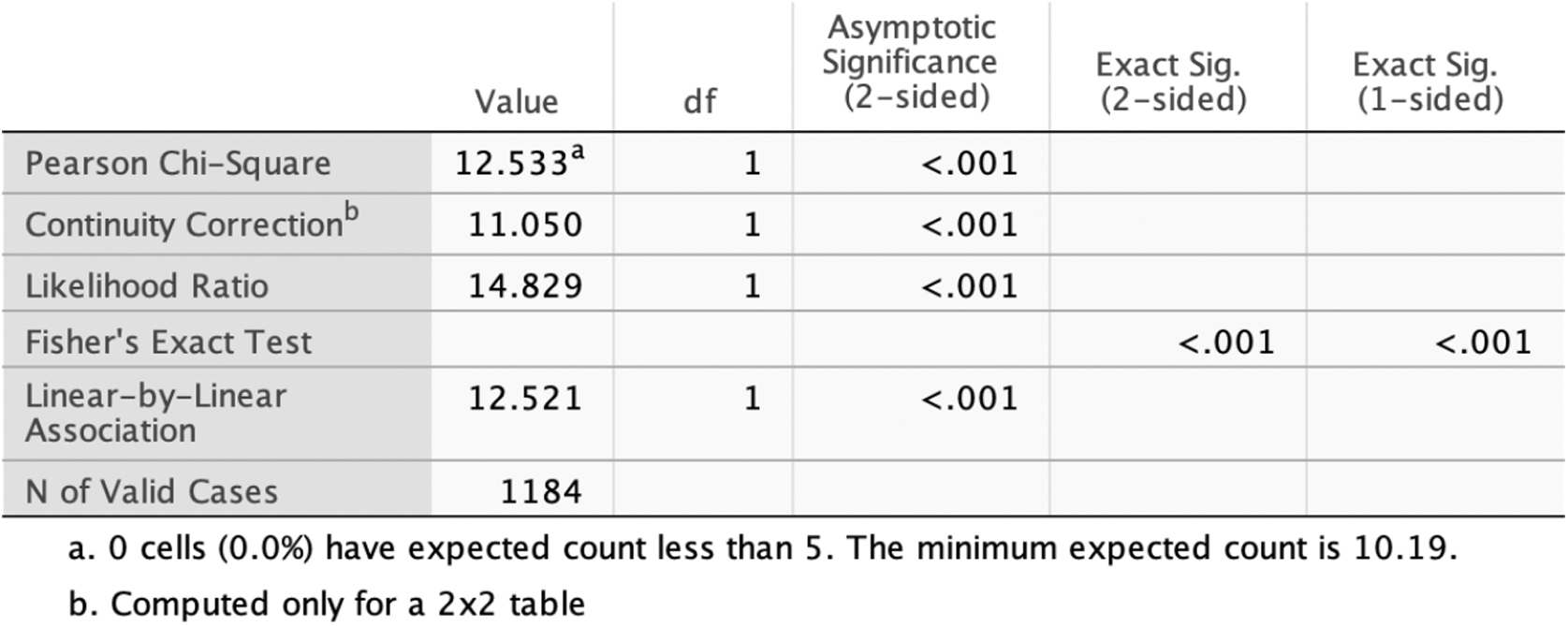
Chi-Square test (p-value of <0.001) shows the significance of the association of ethnicity and heterozygosity.
Discussion
The variations p.G87V and p.Gln298=have known allele frequencies of 0.000007956 and 0.0008988. These numbers are representative of the global population; however, our research indicates that the Mizrahi Jewish populace has significantly elevated allele frequencies. In a previous validation study, 162 DNA specimens of Iranian Jewish were sequenced for LIPA variations. For LIPA p.G87V (ggc>gtc, alternative numbering p.G66V), a heterozygous frequency of 5/162 (3.086%) was discovered. This could be compared to the 24/1184 (2.03%) found in this study. The previous study was done solely in the Mizrahi Jewish community and had not analyzed samples from other Middle Eastern backgrounds. In this study, we included samples from various Middle Eastern groups to investigate if the LIPA variations are more prevalent among Mizrahi Jews compared to other populations within the same demographic. Our results illustrated how these two variants exhibit hardy Weinberg equilibrium, suggesting this ratio should be constant from generation to generation outside of any external factors. The statistical test of chi-square was chosen as it determines the significance of the association between two variables. This test yielded a p-value of <0.001 which confirms that ethnicity and heterozygosity are statically significant.
A larger sample size conveys that more individuals carry the variant, but among the subsets, the Mizrahi Jewish subpopulation overwhelmingly demonstrated an elevated prevalence of LIPA variations compared to other Middle Eastern groups. In the Mizrahi Jewish sample pool of 660, 22 exons 4 and exon 8 LIPA variants were identified, conferring a carrier frequency of 1 in 30, which translates to a LAL-D positive prevalence of 1 in 900 among Mizrahi Jews. This is about 4.6 times higher than the 1 in 4200 previously reported. However, one limitation of this study is the sample size which hinders the applicability and accuracy of suggested prevalence rates. Although the sample size remains less than ideal, this study suggests the Mizrahi Jewish population may carry a greater risk of LAL-D when compared to other Middle Eastern populations. Due to the observed frequency of WD in this subset of the population and the clinical severity of the disease, early detection of variants through genetic testing are essential for the early detection and prevention of clinical disease. Carrier testing for LIPA variants among at-risk communities as well as prenatal testing for at-risk pregnancies is readily available. As such, if an individual has been identified to carry the variation, genetic testing could be performed to prevent significant complications associated with LAL-D. In conclusion, this study elucidates the profound need for genetic screening and early identification of LIPA gene exon 4 variation p.G87V and exon 8 variation p.Gln298=in Middle Eastern populations that maintain an elevated risk of carrying the variation and developing WD or CESD.
Footnotes
Acknowledgment
We appreciate Alexion Pharmaceuticals subsidiary of AstraZeneca for supporting this study.
Authors' Contributions
J.J.: Writing—original draft, data curation. J.F.: Conceptualization, writing—review, and editing. R.T.: Writing—review and editing, methodology. K.Y.: Writing—review and editing. E.B.: Writing—review and editing. E.D.: Writing—review and editing. T.B.: Writing—review and editing. P.H.: Writing—review and editing. S.B.: Supervision, visualization. M.P.: Supervison, formal analysis, methodology. Y.V.: Ayoub—supervision, writing—review and editing, funding acquisition.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.