
Abstract
Background:
Retinitis pigmentosa (RP) is a complex inherited and progressive degenerative retinal disease. The eyes shut homolog (EYS) is frequently associated with RP is surprisingly high. Exploring the function of EYS is quite difficult due to the unique gene size and species specificity. Gene therapy may provide a breakthrough to treat this disease. Therefore, exploring and clarifying pathogenic mutations of EYS-associated RP has important guiding significance for clinical treatment.
Methods:
Clinical and molecular genetic data for EYS-associated RP were retrospectively analyzed. Sanger sequencing was applied to identify novel mutations in these patients. Candidate pathogenic variants were subsequently evaluated using bioinformatic tools.
Results:
A novel pair of compound heterozygous mutations was identified: a novel stop-gain mutation c.2439C>A (p.C813fsX) and a frameshift deletion mutation c.6714delT (p. P2238fsX) of the EYS gene in the RP family. Both of these mutations were rare or absent in the 1000 Genomes Project, dbSNP, and Genome Aggregation Database (gnomAD). These two mutations would result in a lack of multiple functionally important epidermal growth factor-like and Laminin G-like coding regions in EYS.
Conclusions:
A novel compound heterozygote of the EYS gene in a Chinese family with an autosomal inheritance pattern of RP was identified. Identifying more pathogenic mutations and expanding the mutation spectrum of the EYS gene will contribute to a more comprehensive understanding of the molecular pathogenesis of RP disease that could be gained in the future. It also could provide an important basis for the diagnosis, clinical management, and genetic counseling of the disease.
Introduction
Retinitis pigmentosa (RP, OMIM 268000
The most common initial symptoms of the disease include night blindness and progressive visual field loss, which ultimately results in irreversible blindness (Chen et al., 2015; Daiger et al., 2013). The pathogenesis of RP has yet to be completely elucidated (Yang et al., 2020) and over 60 causal genes are registered on the Retinal Information Network database, which demonstrates the genetic heterogeneity of the disease (MIM268000; RetNet).
Many genes relating to RP have been identified, such as ABCA4, RPE65, CERKL, CNGA1, TULP1, PDE6 (PDE6A, PDE6B, PDE6G), and eyes shut homolog (EYS) (Huang et al., 2016; Tsang and Sharma, 2018; Zhang et al., 2018).
The inheritance patterns of RP can be classified as autosomal dominant (30-40% of RP patients), autosomal recessive (AR) (50-60% of AR-RP patients), and X-linked (5-15% of RP patients) (Bunker et al., 1984; Xiao et al., 2017). Approximately 5-10% of AR-RP patients carried mutations of EYS (Littink et al., 2010), whereas the disease in just 1-2% of AR-RP patients was caused by other related genes (Huang et al., 2010; Sengillo et al., 2018; Tsang and Sharma, 2018).
Therefore, EYS-associated RP disease with a high incidence rate has a significant clinical research value. In addition, EYS-associated RP is a disease that progresses incredibly rapidly, and treating it can be more difficult to treat than other AR genes, such as USH2A and MAK (McGuigan et al., 2017). Therefore, the exploration and clarification of the disease mechanism of EYS-associated RP have important guiding significance for clinical treatment.
EYS is encoded by one of the largest eye-specific genes and it was first identified in 2008 (Abd El-Aziz et al., 2008). The EYS gene locus on chromosome 6q12 with over 2 Mb base is capable of encoding an extracellular protein of 3165 amino acids (Abd El-Aziz et al., 2008). EYS protein contains two types of structural domains with unknown functions: Epidermal growth factor (EGF)-like and Laminin G-like domain (Collin et al., 2008).
Although the EYS-associated RP disease has been studied for more than a decade, it has proven difficult to find clinical treatments from the perspective of EYS-protein function exploration. The large gene size and lack of suitable animal models are the main reasons for the research lag. The main reasons for the research lag are large gene size and a lack of suitable animal models. This is due to the fact that with the exception of some primates, such as humans and macaques, other mammals, such as mice, have lost the EYS gene.
Gene replacement therapy is the latest treatment for a variety of ophthalmic diseases, but this cannot be used in EYS-associated RP. The EYS exceeds the 4.7 kb, carrying capacity of the adeno-associated viral (AAV) vectors. EYS-associated RP cannot be treated using AAV gene replace therapy. Site-specific gene correction therapy is a potential breakthrough for curing this disease (Dotzler et al., 2021).
Therefore, the identifications and genetic characterizations of the EYS variants are important for developing EYS-associated RP therapies. However, among the Chinese population with a high incidence of AR-RP (1/3000), EYS mutations and genotype-phenotype correlations have not been well documented (Wei et al., 2020). As a result, a powerful and efficient strategy or platform for RP mutation screening must be established as a means of discovering the putative novel disease causative genes and increasing the possibility of identifying genetic causes for RP patients.
A targeted Whole Exome Sequence (WES) method was utilized in this study for assessing pathogenic variants in the AR-RP gene family among Han Chinese patients. A novel pair of compound heterozygous mutations was identified in this Chinese RP family. Patients in this RP family were found to be carrying a novel stop-gain mutation c.2439C>A (p.C813fsX) and a frameshift deletion mutation c.6714delT (p. P2238fsX). Both mutations are rare or absent from the 1000 Genomes Project, dbSNP and Genome Aggregation Database (gnomAD). In addition, the novel mutation has not been previously reported in AR-RP patients. The findings of this study will serve to the EYS mutation spectrum in AR-RP patients.
Materials and Methods
Study subjects and ethics statement
For this study, a Han Chinese family with serious RP was recruited for this study. Written informed consent was obtained from every patient and family members. The study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Ethics Review Boards of Sichuan Provincial People's Hospital. All experiments were performed in accordance with the approved protocols.
Clinical diagnosis
A complete ophthalmic examination, including visual acuity, fluorescence fundus angiography, optical coherence tomography (OCT), visual field test, and full-field electroretinogram (ERG), was performed on each volunteer and peripheral blood samples were collected from all study subjects. Ophthalmologists at the Sichuan Academy of Medical Sciences and Sichuan Provincial People's Hospital assessed the clinical data.
DNA extraction
Genomic DNA was extracted from peripheral blood samples that were obtained from RP patients and their unaffected relatives. Based on the principle that cetyltrimethylammonium bromide (CTAB) forms complexes with proteins and polysaccharide, while not precipitating nucleic acids, ethanol precipitation was added for the purpose of completely separating the genomic DNA completely after extraction with organic isopropanol solvent to remove protein, polysaccharide, and phenolic impurities.
Whole-exome sequencing and Sanger sequencing
The genomic DNA samples of individuals II-3, IV-1, and III-2 from the RP family were subjected to WES analysis. Sequencing libraries were prepared using the SureSelect XT Target Enrichment Kit (Agilent Technologies, Santa Clara, CA) and captured using the Agilent Sure Select Human All Exon Kit V5 (Agilent Technologies). Paired-end sequencing was conducted using the HiSeq 2500PE100 platform (Illumina, San Diego, CA) with a read length of 100 bp and an average coverage depth of a minimum of at least 100 × for each sample.
Sanger sequencing was used to verify identified variants of EYS. All exons were amplified from the genomic DNAs by PCR using a standard condition before being sequenced on a 3730 ABI DNA sequencer (Thermo-Fisher). The sequencing results were then ultimately analyzed using ApE (A plasmid Editor) software.
Mutation identification and analysis
Close attention was paid to the functional SNPs/Indels in homozygous or compound heterozygous variants, including frameshift insertions or deletions, non-synonymous variants, and splicing junction variants with a greater likelihood of being pathogenic to identify the disease-causative variant in the family with RP. Common variants with high-frequency present were filtered in dbSNP138, 1000 Genomes Project, Exome Aggregation Consortium, OMIM, HGMD, and other East Asian population databases.
The genes that were affected by these filtered variants were then compared with the reported genes associated with retinal diseases. The Burrows-Wheeler Aligner program was used to align the sequencing reads to the reference human genome assembly hg38 UCSC. The “mpileup” in SAMTOOLS v.0.1.19 command was applied for identifying insertions/deletions and single nucleotide variants (Tian et al., 2020). The NCBI CCDS, RefSeq, and EnSembl databases were referred for filtering and annotating the detected variants.
The WES results from patients with AR-RP were filtered as follows: (1) variants in intergenic, intronic, and untranslated regions were excluded; (2) synonymous variants were excluded without altering splice-site regions being altered; and (3) variants with minor allele frequencies that were ≥0.01 in any of the three public population databases Exome Aggregation Consortium (ExAC), 1000 Genomes Project (1000G), and gnomAD public population databases were excluded.
Mutations identified in this study were verified in the OMIM and HGMD databases to confirm whether the pathogenic mutations had been previously reported. Co-segregation analysis was performed for all family members after the above filtering steps, and variant scores that corresponded to the quality of biological and statistical evidence were ultimately assigned. Strong candidates were validated by Sanger Sequencing.
All identified variants were assessed using the following online tools and databases. HomoloGene (https://www.ncbi.nlm.nih.gov/homologene) from the NCBI website was used for evaluating the amino acid conservation of EYS. The structural model of the EYS protein was downloaded in RCSB PDB, and the SWISS website was used for constructing construct wild-type and mutant structural models.
PyMOL was used to predict effective amino acid mutations with no protein structure. In addition, online bioinformatics tools, such as the PROVEAN program, Sorting Intolerant from Tolerant (SIFT), and MutationTaster, were used for predicting the potential pathogenic effect amino acid substitution in EYS.
Results
Clinical characteristics
This study recruited a three-generation Chinese family with RP, and no consanguineous marriage history was recruited for this study (Fig. 1). The proband (III:2) in this family was a female with congenital night blindness and progressive peripheral vision loss. A pattern of AR inheritance in the family was identified by pedigree analysis and this consisted of eight affected and eight unaffected members.
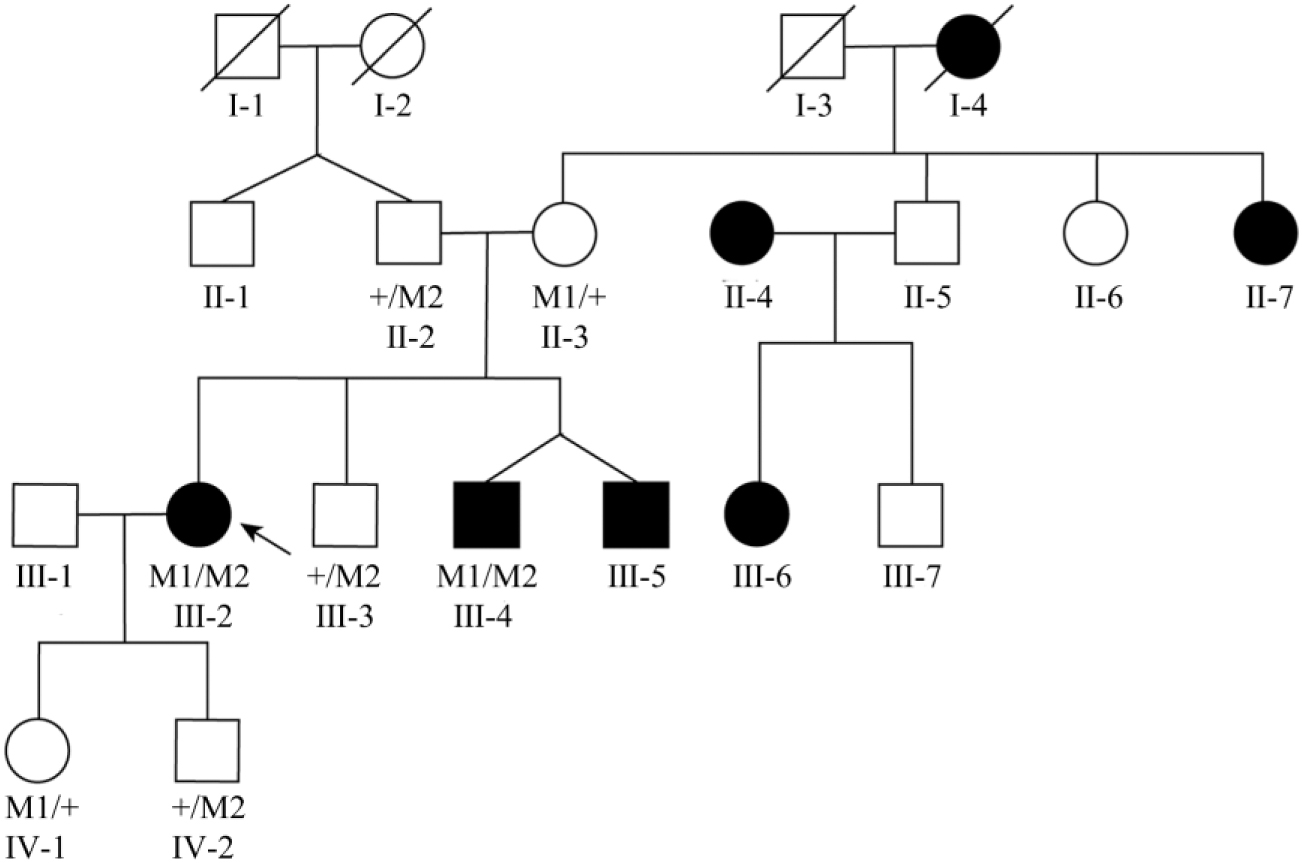
Pedigree of the Chinese family with RP. The desegregation of compound heterozygous changes c.2439C>A (M1) and c.6714delT (M2). Genotypes are presented as follows: M1/M2 represents individuals with both mutations as compound heterozygous; M1/+ and +/M2 indicate heterozygous carriers; and +/+ indicates individuals carrying two wild-type alleles. Unlabeled indicates unobtainable DNA samples. The arrow points to the proband. The female and male are indicated by a circle and a square, respectively. Empty and filled symbols represent normal and affected individuals, respectively. RP, retinitis pigmentosa.
Fluorescence fundus angiography discovered slender retinal vessels in both eyes of the proband, no obvious retinal vessels in the middle-periphery of each quadrant, extensive retinal pigment epithelium (RPE) changes in the posterior pole and the middle-periphery of each quadrant, more osteocyte-like pigment masking fluorescence impression in the middle and periphery of each quadrant, and RP in both eyes of the proband (Fig. 2A).
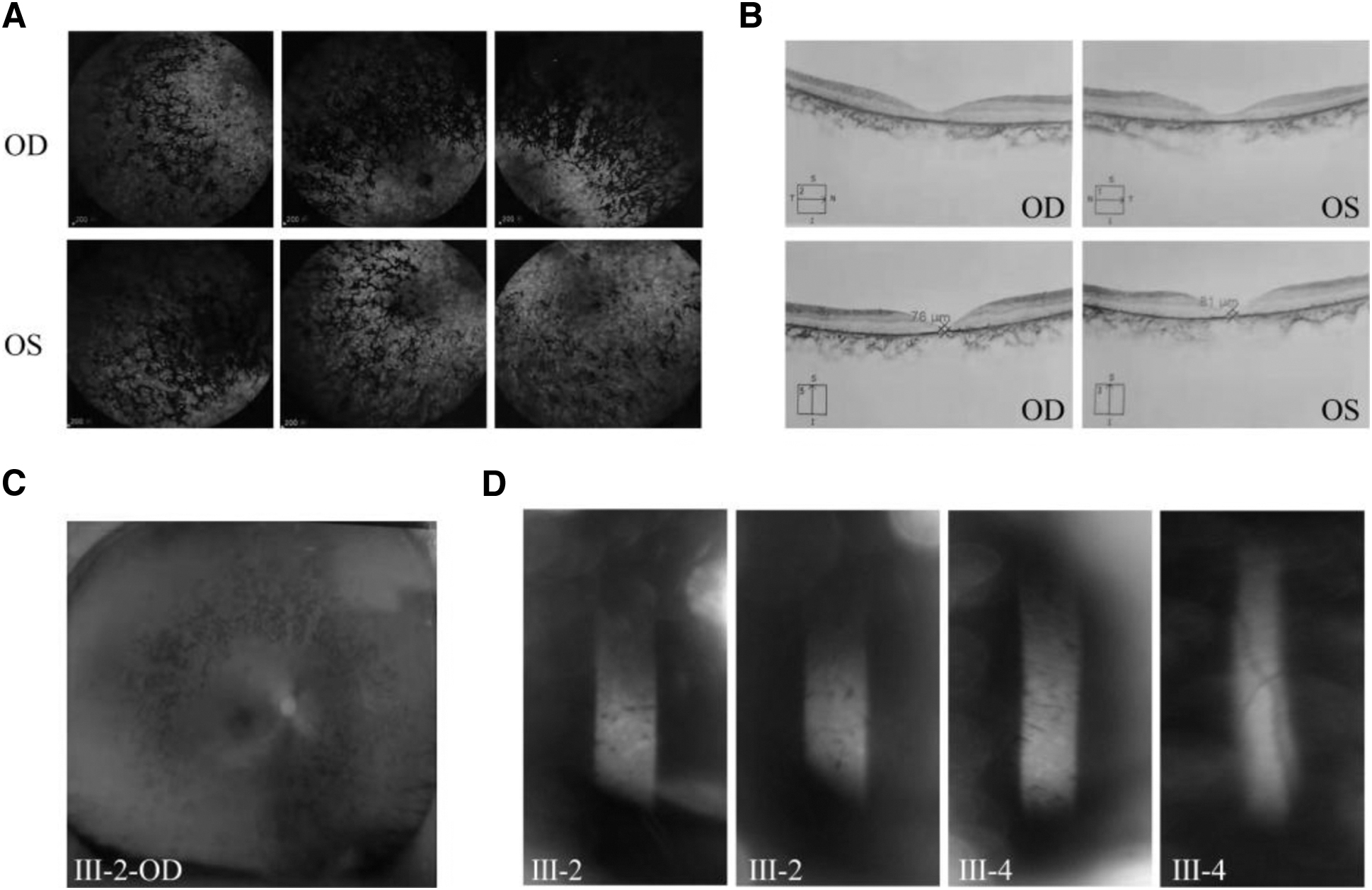
Clinical data of the proband of family AR-RP.
Fundus images identified pigment deposition (Fig. 2C). OCT found thinned macular fovea retina (Fig. 2B), and the visual field test suggested a loss of peripheral vision and only had a central tubular visual field in both eyes (Fig. 2D). The ERG analysis found there to be no response to the stimulus for both cone and rod photoreceptors.
Pattern visual evoked potential found that the waveform of both eyes was not typical. The latency of P100 in the right eye was almost normal, whereas the amplitude of P100 decreased significantly. Conversely, the latency of P100 in the left eye was delayed, whereas the amplitude of P100 decreased significantly.
The patient was classified as a congenital RP. Relevant clinical test data are shown in Tables 1-4. It was also found that two brothers of the proband (Ⅲ:4, Ⅲ:5) had the same phenotype and were subsequently diagnosed with congenital RP at an early age. In contrast, the fundus photograph was normal among unaffected individuals who were unaffected.
Clinical Characters Data of Patients with Retinitis Pigmentosa
—, mean unknown; MD, mean defect; OD, right eye; OS, left eye.
Electrophysiological Measurement and Data of Electroretinogram in the Proband
ERG, electroretinogram.
Electrophysiological Measurements of Pattern-Visual Evoked Potential in the Proband
Ophthalmic Audition and Optometry Results
A, axial position; C, cylindrical degree; PD, pupil distance; S, spherical degree; VD, eye distance.
Verification of variants in EYS
DNA samples were collected from the brothers and parents of the proband for Sanger sequencing as a means of validating this mutation. The father and brother Ⅲ:3 were found not to be carrying the c.2439C>A (p.C813X) mutation, whereas the mother and brothers Ⅲ:3 and Ⅲ:4 had the c.2439C>A mutation.
This novel mutation had not been previously reported in RP patients and was also not in our in-house database of 1000 ethnicity-matched control samples. At the same time, another reported heterozygous mutation, c.6714delT (p.P2238fs), was identified in the proband and their affected sibling.
This is in accordance with the AR phenomenon of co-segregation and compound heterozygosity of genotype and phenotype of the family when the c.2439C>A mutation and c.6714delT mutation are also carried. The Sanger sequencing results for all samples are shown in Figure 3.
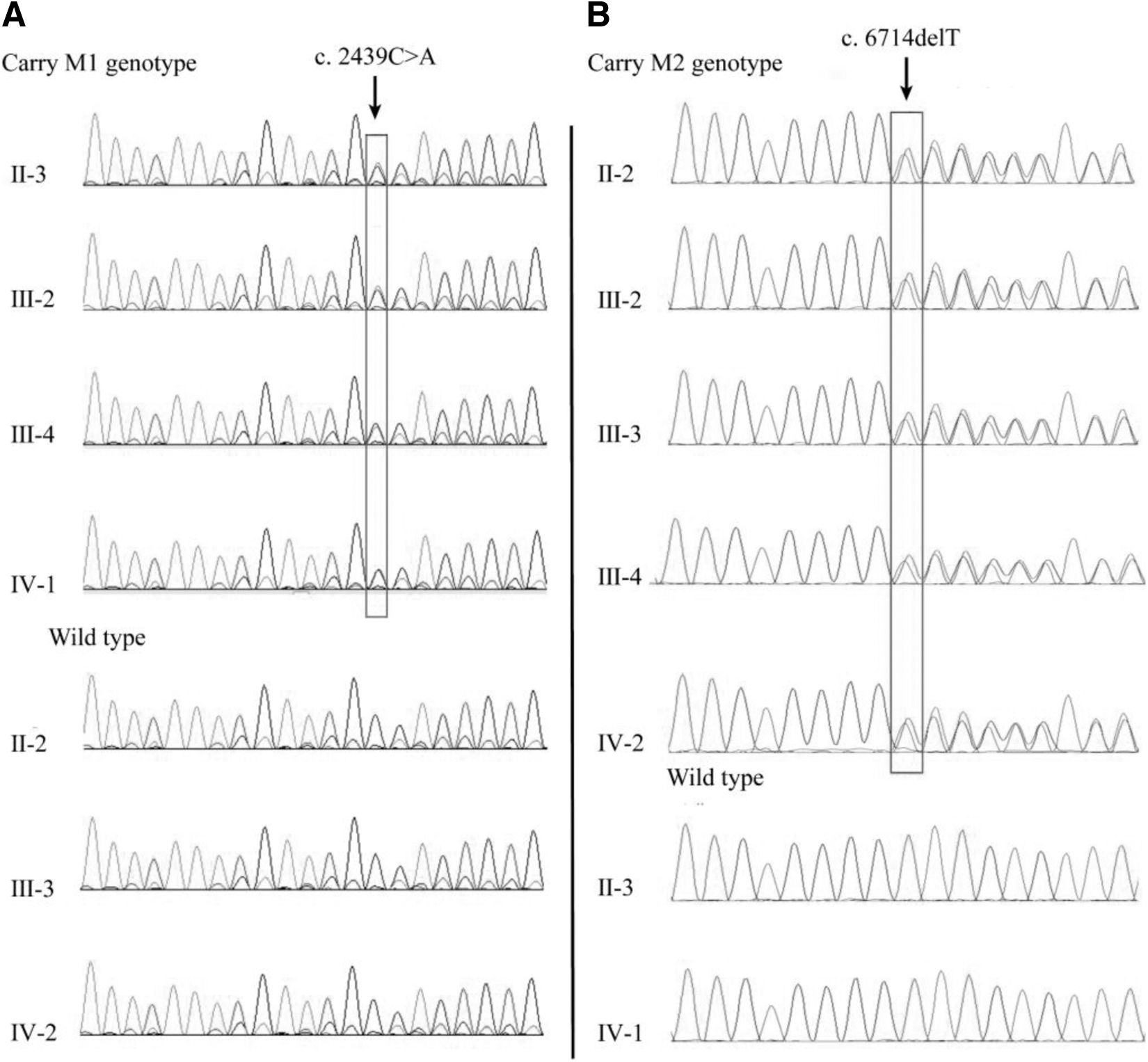
The result of sanger sequence.
In silico analysis of the variants detected in EYS
In silico analyses by PROVEAN program, SIFT, and MutationTaster bioinformatics programs found that the identified EYS variant was predicted to be deleterious (Table 5). According to the American College of Medical Genetics and Genomics (ACMG) guidelines, the stop-gain mutation c.2439C>A (p.C813X) identified that this study will likely be a causative mutation.
Mutation Characteristics and Pathogenic Effect Analysis
D, deleterious; NA, not available.
This will result in premature truncation within the 11th EGF-like domain, which means that the mutant protein will lack 18 EGF-like domains and 5 Laminin G-like domains. The amino acid residues that are affected by the mutation identified in this study are highly conserved in vertebrates, indicating the importance of these residues (Fig. 4A, B).
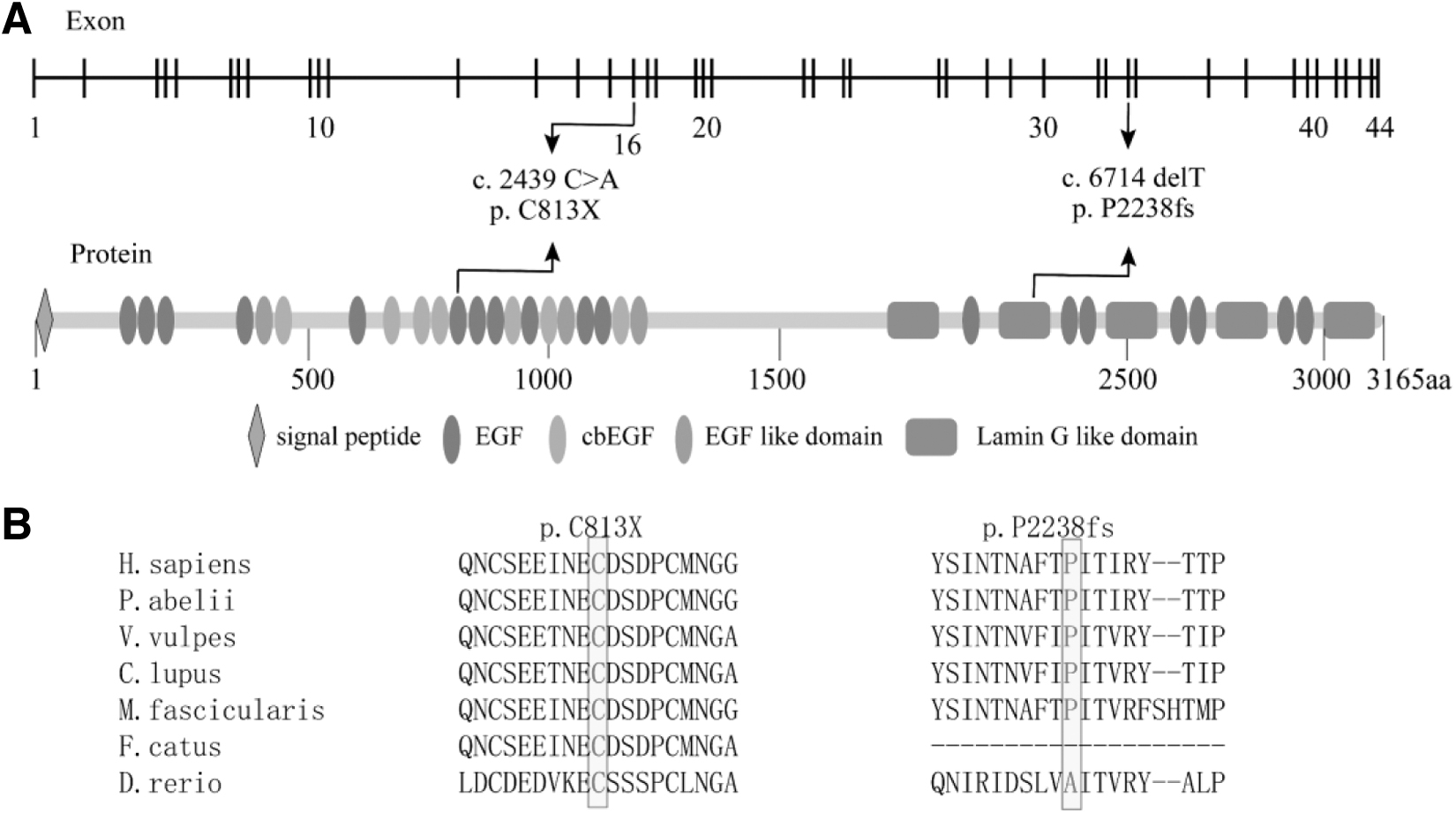
EYS mutations in protein domains and sequence alignment of the affected amino acid residues.
Known heterozygous mutation c.2439C>A (p.C813X) will also result in the loss of six EGF-like and three Laminin G-like domains. Due to the absence of these functionally important domains, the truncated proteins may have little or no remaining function.
Discussion
A pair of compound heterozygous mutations of the EYS gene was identified in a Chinese RP family by this study: c.2439C>A (p.C813fsX) and c.6714delT (p.P2238fsX). These mutations may result in a shortening protein and an abnormal function. Two variations of the proband are inherited from her parents, who only carry one heterozygous variation.
This is consistent with the AR inheritance mode. The heterozygous mutation c.6714delT had been reported in another RP family (Collin et al., 2008), and mutation c.2439C>A had not been previously reported. The two mutations are not polymorphic, and there is a low frequency of occurrence in the population. As shown in Table 6, both mutations were rare or absent in the 1000 Genomes Project, dbSNP and gnomAD.
Frequency of Mutations in Databases
gnomAD, Genome Aggregation Database; MAF, minor allele frequencies.
The comprehensive mining of mutation sites can be useful for the determination and exploration of EYS-associated RP gene therapy treatment. Therefore, the pair of new compound heterozygous mutations in this study identified have important clinical diagnostic significance.
Bioinformatics predicted that these mutations may affect EYS function, so the potential impact of mutations on EYS was also explored. The expression of EYS is rare and specific in different mammals. It was previously believed that this is specifically expressed in the photoreceptor cell layer of the retina, which is only essential for the development and morphology of photo-receptors (Abd El-Aziz et al., 2008).
However, researchers have recently noticed that zebrafish have the ability to express the EYS. Therefore, an important breakthrough was made in EYS biological function. It had been reported that EYS-deficient zebrafish exhibit photoreceptor degeneration, thereby confirming the role played by EYS in photoreceptor survival (Lu et al., 2017; Messchaert et al., 2018; Yu et al., 2016).
Numerous subsequent studies have indicated EYS protein to be highly enriched near the connecting cilium/transition zone as well as in the cytoplasm of the ganglion cells (Alfano et al., 2016). All of these studies suggest the involvement of EYS to be involved in a variety of ocular biological functions, including cellular metabolism, phototransduction cascades, cell signaling, RNA and protein modification, and phagocytosis (Iwanami et al., 2012).
Therefore, the functional loss of protein caused by EYS mutations has an irreparable negative effect on the normal biological function of the eye.
In all the functional studies of EYS, the effect of protein structure change on function has been examined. EYS protein encoded by this transcript consists of 3165 amino acids and is secreted into the extracellular environment due to the fact that it contains a signal peptide. This protein harbors 28 EGF-like and 5 Laminin G-like domains, which determine EYS activity (Collin et al., 2008).
The EGF-like domain and the Laminin G-like domain both have ability to mediate protein-to-protein adhesion through binding, meaning that they participate in visual signal transduction, adhesion, migration, and differentiation processes (Ma et al., 2005; McClung et al., 2012; Messchaert et al., 2018). EGF domain-containing proteins are important ocular tissue development regulators that are found in many proteins.
Low-density lipoprotein receptors and thrombin, which determine important eye functions, contain an abundance of EGF and EGF-like domains. The biological activities of these two proteins are generally dependent on the binding state of the module on the EGF/EGF-like domain to Ca2+.
When a mutation occurs in the EGF module, this leads directly to protein inactivation (Stenflo et al., 2000). CRB1 protein is another important protein in the eye and also has functional EGF/EGF-like domains (den Hollander et al., 1999). Previous literature reported that destroying the 19th cbEGF domain on the CRB1 protein affected the binding of the protein to Ca2+, which ultimately destroys the structural integrity of protein CRB1 in the retinal photoreceptor matrix, which resulted in the loss of retinal photoceptor function.
It can be concluded that the integrity of EGF/EGF-like domain can determine the biological activity of EYS, and the defect of this domain may be the core cause of eye disease.
The regular operation of the EYS protein function operation can generally be guaranteed by the existence of EGF-like and Laminin G-like domains. In this study, the newly discovered stop-gain mutation c.2439C>A (p.C813fsX) was found to disrupt the translation of codon 813 and produced a truncated protein. This mutation results in premature truncation in the 11th EGF-like domain, which means that the mutant protein will lack 18 EGF-like domains and 5 Laminin G-like domains.
Moreover, c.6714delT (p.P2238fsX) will lose six EGF-like and three Laminin G-like domains. As mentioned earlier, it is speculated that the functional defect of the EGF-like domain may potentially cause the loss of the protein and calcium ion binding sites, whereas the failure of all Laminin G-like domain structures will cause the direct loss of all protein-to-protein sites in the EYS protein, thereby impacting the function of the protein.
In conclusion, this study identified a novel pair of compound heterozygous mutations in AR-RP patients using a targeted WGS approach. It expanded the mutation spectrum of the EYS gene in the Chinese population. The findings of this study should be useful for improving the understanding of the molecular pathogenesis of AR-RP disease for diagnosis, clinical management, and genetic counseling purposes.
Footnotes
Acknowledgments
The authors thank the patients and their family members for their invaluable participation in the study.
Authors' Contributions
All the authors have contributed to this study. The concept and design of the study were performed by Y.S. and L.J.; the material preparation, data acquisition, and analysis were performed by Y.W., W.R., C.X., S.D., and Q.L.; and the first draft of the manuscript was written by C.D., which all the authors revised. All authors have approved the final version and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors have seen and approved the submission and take full responsibility for the manuscript.
Data Availability
The research data included in this article can be obtained from the corresponding authors on reasonable request.
Ethics Approval and Consent to Participate
The study adhered to the tenets of the Declaration of Helsinki and received approval from the Institutional Ethics Review Boards of Sichuan Provincial People's Hospital. Support for this case-control association study was provided by the Institutional Review Board of Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital in Sichuan Province, China. All experiments were performed in accordance with approved protocols. Informed consent was obtained from all individual participants included in the study.
Consent for Publication
The authors affirm that human research participants provided informed consent for publication. All participants agreed to publish their medical information.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (Nos. 82271120 and 82121003 to Y.S., No. 82201234 to L.J.), the CAMS Innovation Fund for Medical Sciences (2019-12M-5-032), Sichuan Science and Technology Program (No. 2022ZYD0066 to Y.S., 2022YFS0606 to L.J., and 2023YFS0086 to C.X.).