
Abstract
Objective:
To investigate the association between ACTN4 gene mutation and primary nephrotic syndrome (PNS) in children in Guangxi Autonomous Region, China.
Methods:
The high-throughput sequencing technology was used to sequence ACTN4 gene in 155 children with PNS in Guangxi Autonomous Region in China, with 98 healthy children serving as controls. Twenty-three exon-specific capture probes targeting ACTN4 were designed and used to hybridize with the genomic DNA library. The targeted genomic region DNA fragments were enriched and sequenced. The protein levels of ACTN4 in both case and control groups were quantified using ELISA method.
Results:
Bioinformatics analysis revealed five unique ACTN4 mutations exclusively in patients with PNS, including c.1516G>A (p.G506S) on one exon in 2 patients, c.1442 + 10G>A at the splice site in 1 patient, c.1649A>G (p.D550G) on exon in 1 patient, c.2191-4G>A at the cleavage site in 2 patients, and c.2315C>T (p.A772V) on one exon in 1 patient. The c.1649A>G (p.D550G) and c.2315C>T (p.A772V) were identified from the same patient. Notably, c.1649A>G (p.D550G) represents a novel mutation in ACTN4. In addition, three other ACTN4 polymorphisms occurred in both case and control groups, including c.162 + 6C>T (1 patient in case group and 2 patients in control group), c.572 + 11G>A (1 patient in case group and 2 patients in control group), and c.2191-5C>T (4 patients in the case group and 3 patients in control group). The serum ACTN4 concentration in the case group was markedly higher, averaging 544.7 ng/mL (range: 264.6-952.6 ng/mL), compared with 241.20 ng/mL (range: 110.75-542.35 ng/mL) in the control group.
Conclusion:
Five ACTN4 polymorphisms were identified among children with PNS in Guangxi Autonomous Region, China, including the novel mutation c.1649A>G. The lower serum levels of α-actinin-4 in the case group suggest that this protein might play a protective role in PNS.
Introduction
Nephrotic syndrome is a clinical syndrome with a series of pathophysiological changes due to the increased permeability of glomerular filtration membrane to plasma protein and the loss of a large amount of plasma protein from urine (Wang et al., 2015). Primary nephrotic syndrome (PNS) is a type of immune-mediated glomerular disease (Yokoyama et al., 2015), which has severe effects on the normal growth and development of children and their quality of life. In terms of clinical characteristics, PNS is usually classified into five categories as follows: focal segmental glomerulosclerosis (FSGS), minimal change nephropathy, IgA nephropathy, membrane nephropathy, and without renal puncture (Wang et al., 2015; Yokoyama et al., 2015). The research on pathogenic genes associated with PNS has come to attract attention, since these genes play an important role in the development of PNS (Liu and Wang, 2017). The ACTN4 gene is located on the long arm of chromosome 19 (NC_000019.9, chr19:39138289-39222229) and composes of 23 exons. The ACTN4 mutations are a cause of autosomal-dominant FSGS (Kaplan et al., 2000). It has been demonstrated that mutant forms of this gene are usually associated with abnormal serum levels, especially in patients suffering from FSGS (Kaplan et al., 2000).
It is conceivable that a higher number of PNS patients harbor mutations in the ACTN4. In order to investigate the relationship between ACTN4 mutations and PNS, we conducted a screening and sequencing study of peripheral blood samples from 134 young PNS patients with 107 healthy controls. This analysis utilized next-generation sequencing technology in conjunction with the FastTarget target gene capture method.
Materials and Methods
Study subject
A total of 134 cases with PNS were recruited from the outpatient clinics and wards of the Affiliated Hospital of Youjiang Medical College for Nationalities from July 2015 to September 2017. All these patients were under 16 years of age, and those who were diagnosed with nephrotic diseases or had secondary nephrotic syndrome were excluded. During the same period, a total of 107 healthy children were enrolled as controls. These children had no family history of the nephrotic syndrome or other kidney disease. All the subjects were from Guangxi Zhuang nationality for at least three generations and were not related to each other.
This research was approved by the Ethics Committee of the Affiliated Hospital of Youjiang Medical College for Nationalities. Written informed consents were signed by all the parents/guardians before the study and they were aware of the study purpose, risks, and benefits.
The main equipment and reagents
AB 2720 Thermal Cycler (Applied Biosystems, Waltham, MA); Eppendorf 5810R Centrifuge (Eppendorf, Hamburg, Germany); NanoDrop (NanoDrop Technologies, Wilmington, DE); Invitrogen Qubit Spectrophotometer (Invitrogen, Carlsbad, CA); Illumina NextSeq (Illumina, San Diego, CA, USA); Multiskan FC Microplate reader (Thermo Fisher Scientific); Micropipettes (Eppendorf, Germany); Nextseq Reagent KitV2 (Illumina, San Diego, CA); TIANGEN Gel Extraction kit (TIANGEN, Beijing, China); 10×Reaction buffer (TaKaRa, Dalian, China); HotStarTaq polymerase (TaKaRa, Dalian, China); ACTN4 ELISA Kit (Enzyme linked biology, China); and DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany).
Genomic DNA extraction
Genomic DNA was extracted from the 2 mL peripheral blood samples collected from fasting children using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s standard protocol. The purity of the extracted DNA was assessed by measuring the ratio of absorbance at 260 nm to absorbance at 280 nm using the Invitrogen Qubit Spectrophotometer.
FastTarget target region sequencing
In order to obtain the sequence of the target region and perform sequencing, the human genome assembly UCSC hg19 (version GRCh37) was used as the template in primers design designing primers for sequencing the ACTN4 target region. The primers were selected and optimized to obtain a clear single band of sequence. A total of 20 pairs of optimized primers were mixed thoroughly into multiplex PCR primer panels according to the protocol, and the quality control was implied. Based on the special method of capillary electrophoresis, the final number of optimized panels obtained is about one twentieth of the number of primers. We used primers incorporating index sequences to introduce Illumina-compatible tags to the ends of the library by PCR amplification. A 11-cycle PCR program was used in this reaction to minimize the propensity of products. A FastTarget sequencing library was produced by mixing the amplified products with buffer and tapping them with a syringe. The fragment lengths were determined using the Agilent 2100 Bioanalyzer. The prepared fragments were subjected into the Illumina MiSeq platform, resulting in 2 × 150 bp paired-end sequencing reads. After eliminating adapters and low-quality reads, the clean reads were subjected for further analysis.
Data screening and biological information analysis
We aligned the sequencing data to the UCSC hg19 human genome using BWA software (v0.7) and then were validated using the GATK software (v3.7). To verify the sequence accuracy, an advanced comparison identified SNVs/InDels. The SNVs and InDels in each sample were identified using VarScan software and GATK HaplotypeCaller mode. The abovementioned detection schemes were compared to determine the SNV/InDel, and the samples were combined accordingly. SNV/InDel loci were compared with the dbSNP, thousands of human genomes, ESP6500, ExAC03, ExA C03_EAS, genomAD, and Hrcr1Kaviar_20150923 databases by ANNOVAR to evaluate the frequencies, functional characteristics, conservation of these sites, and pathogenicity and to confirm the most significant SNV/InDel sites for the data obtained.
Determination of serum ACTN4 levels
The protein expression levels of ACTN4 were quantified using a double-antibody sandwich ELISA, according to the kit operating instructions.
Statistical analysis
SPSS 22.0 software was used for the statistical analysis, PLINK analysis software for correlation analysis, and Haploview software for linkage disequilibrium analysis. The association analysis was performed based on genotypic and phenotypic data (sex, age, etc.) of the mutation sites of the existing samples to identify the disease-related loci. The ACTN4 genotypes and alleles between case and control groups were compared, and the Hardy-Weinberg genetic balance test was performed. Logistic regression was performed on a single locus basis with five hypothesized genetic models, Dominant (Low frequency allele is dominant), Recessive (Low frequency allele is recessive), Log-additive (Additive effect), and HOM/HET (codominant model, with homozygous normal reference) for correlation analysis.
Results
Statistic power and Hardy-Weinberg equilibrium test
The statistic power of our analysis was estimated by GPower software (v3.1). The results of the analysis indicated that the statistical power for the IFN-γ rs2069727 analysis was 0.769, whereas it was 0.478 for the rs1861494 polymorphism. The genotype and allele frequencies for the IFN-γ rs2069727 and rs1861494 polymorphisms in the control group adhered to Hardy-Weinberg Equilibrium (HWE), with p-values of 0.970 and 0.205, respectively. This compliance suggests that our study population is representative of the general population.
Sequencing depth and coverage
In the patient samples, 86.50% of the target regions exhibited a sequencing depth exceeding 2x, compared with 98.20% in the control samples. In addition, 76.00% of the target regions in patient samples and 94.70% in control samples demonstrated a sequencing depth greater than 30x. This indicates that our sequencing data are reliable and sufficient to support our subsequent analysis results.
ACTN4 gene mutation
Results of ACTN4 mutation analysis and sequencing showed that five ACTN4 polymorphisms were identified only in PNS patients, including c.1516G > A (p.G506S) on exon 13 in two PNS patients, one with minimal change nephropathy and another without renal puncture; c.1442 + 10G > A at the splice site in a minimal change nephropathy patient; c.2191-4G > A at the cleavage site, identified from two FSGS patients; and c.1649A > G (p.D550G) on exon 14 together with c.2191-4G > A at the cleavage sites, identified from two FSGS patients; and c.2315C > T (p.A772V) on exon18 in one patient.
Two vital mutations were detected in exons, including c.1649A > G (p.D550G) in exon14 and c.2315C > T (p.A772V) in the exon18. These two mutations were observed in an 8-year-old boy and an 11-year-old girl, respectively, both diagnosed with PNS and lacking a familial history of the condition. Bioinformatic analysis confirmed that these mutations are pathogenic, potentially resulting in altered protein expression and subsequent dysfunction.
Potential pathogenic mutations
Analysis of sequencing results revealed 3 ACTN4 polymorphisms that occurred in both case and control groups, including exon1: c.162 + 6C > T heterozygous mutations at the cleavage site (1 in the case group and 2 in the control group), exon5: c.572 + 11G > A heterozygous mutation (1 in case group and 2 in control group), and exon18: c.2191-5C > T heterozygous mutations (4 in case group and 3 in control group) (Table 1, Fig. 1). In addition, another 3 ACTN4 polymorphisms only occurred in control patients, including exon1: c.162 + 9C > T heterozygous mutations at the cleavage site, exon14: c.1691A > G (p.E564G) heterozygous mutation, and exon16: c.1901A > C (p.D634A).
ACTN4 Mutation Results for All Samples at Low Frequency
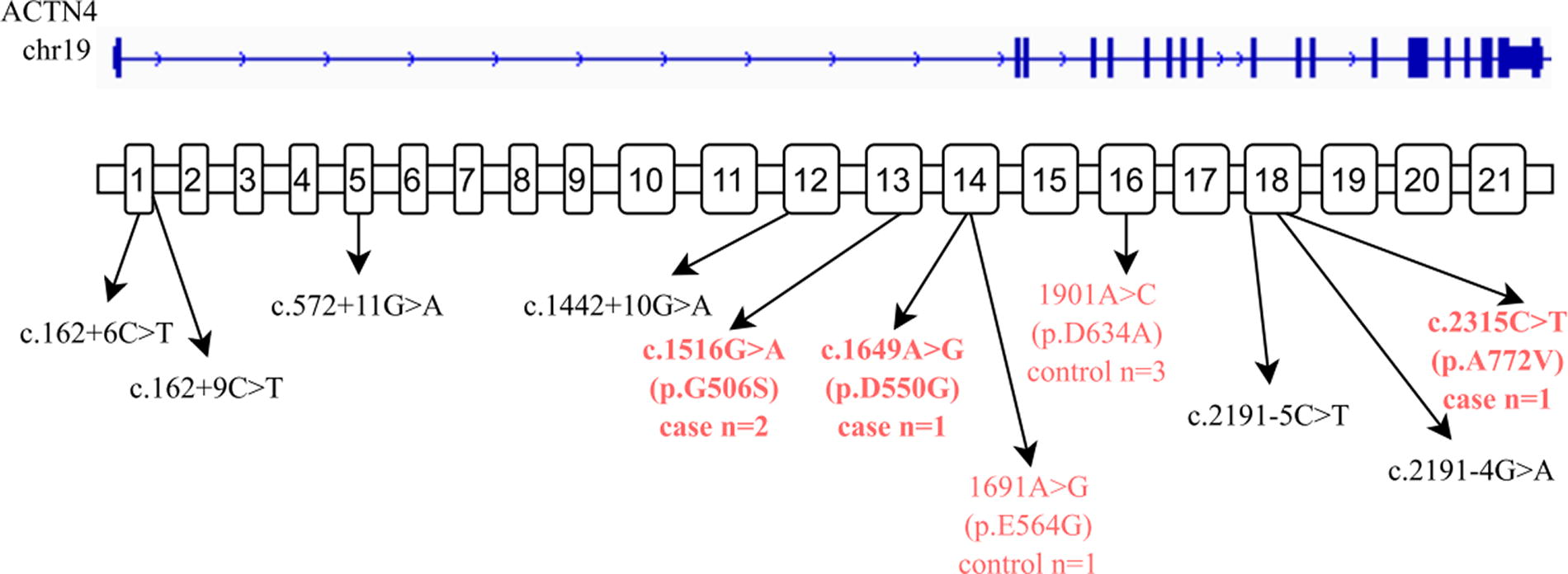
The mutations located in the ACTN4 gene.
Newly identified mutation
Analysis of sequencing results revealed 4 heterozygous polymorphisms, including exon14: c.1649A > G (p.D550G), exon14: c.1691A > G (p.E564G), exon16: c.1901A > C (p.D634A), and exon1: c.162 + 9C > T at the cleavage site. These mutations were newly identified ACTN4 mutations and were not found in the databases of dbSNP, 1000 Genomes, ESP6500, ExAC03, ExAC03_EAS, gnomAD, Hrcr1, Kaviar_20150923, and GENESKYDB_Freq (Table 2).
Probability of All Low-Frequency Samples in the Database
Among them, the exon14: c.1649A > G (p.D550G) heterozygous mutation was only detected in case group, whereas exon14: c.1691A > G (p.E564G) heterozygous mutation, exon16: c.1901A > C (p.D634A) heterozygous mutation, and exon1: c.162 + 9C > T heterozygous mutation at the cleavage site were only found in the control group.
SNP association analysis
All SNPs were examined using codominant, dominant, recessive, and allele models. As shown in Table 3, significant differences were observed between the case and control groups for rs78078603 in both the codominant (χ2 = 9, p = 0.011) and dominant models (χ2 = 3.909, p = 0.048) and for rs79072548 in the codominant (χ2 = 15.26, p = 0.0005), dominant (χ2 = 13.91, p = 0.0002), and allele-specific (χ2 = 14.34, p = 0.0002) models.
SNP Site Chi-Square Test Results
FDR_BH_adjusted, FDR corrected p value.
Logistic regression analysis of SNP loci
A comprehensive logistic regression analysis of all SNP loci was conducted, with adjustments made for gender and age to control for their potential confounding effects (Table 4). We explored various genetic models to understand the influence of allele frequency and type on disease association. Specifically, we tested the dominant (where the low-frequency allele exerts a dominant effect), recessive (where the low-frequency allele is recessive), log-additive (considering an additive effect of alleles), and codominant (HOM/HET, with homozygous normal as the reference) models to identify correlations with the phenotype.
Logistic Regression Analysis of Environmental Factor Correction for SNP Loci
NMISS, no missing data in this analysis model; OR, logistic regression OR; L95, U95, logistic regression 95% confidence interval; SE, standard error; STAT, t-statistic of the coefficient.
The analysis revealed notable findings regarding the SNP rs3745859. The heterozygous (HET) model showed a significant association (STAT = 2.516, p = 0.01186), indicating that individuals carrying one copy of the risk allele and one copy of the normal allele have a different disease risk compared with those with two normal alleles. This suggests that even a single copy of the variant allele at this locus can influence the trait, which may be indicative of a semidominant genetic effect where the presence of the variant allele alters protein function or expression in a way that impacts the disease phenotype.
In addition, the dominant model demonstrated a significant difference between the case and control groups for this SNP (STAT = 2.378, p = 0.0174). This result supports the idea that the presence of at least one variant allele (either homozygous or heterozygous for the variant) can increase disease susceptibility compared to the homozygous normal genotype.
Similarly, our analyses suggest that the dominant, log-additive, and heterozygous models provide a compelling explanation for the associations observed with SNPs rs78078603 and rs79072548 in relation to primary nephrotic syndrome (PNS). These findings are critical as they highlight the potential mechanisms through which these genetic variations contribute to PNS pathogenesis. For instance, the log-additive model’s significance suggests that each additional copy of the variant allele incrementally increases the risk of PNS, a pattern often seen in polygenic diseases where multiple genes contribute to the condition.
These insights are crucial for understanding the genetic architecture of PNS and could guide future research into targeted therapies or diagnostic markers based on these genetic variations. Further studies are needed to elucidate the biological pathways affected by these SNPs and to validate these associations in larger diverse populations.
Genotype-phenotype association analyses
The phenotypic information of the samples can also be used for genotype association analysis. In the data analysis, we conducted a correlation analysis of all phenotypic information (except sex, age) with genotypes. At the same time, gender and age were used for correction. As shown in Table 5, the rs2112649 in ACTN4 was significantly different in log-additive, recessive, and HOM analysis models.
Genotype-Phenotype Association Analyses
NMISS, no missing data in this analysis model; L95, U95, logistic regression 95% confidence interval; SE, standard error; STAT, t-statistic of the coefficient.
Linkage disequilibrium analysis
In this study, haplotype software was used to analyze the linkage disequilibrium of rs2112649, rs2287728, rs78078603, rs3745859, rs899199, rs62120068, rs12981131, and rs79072548 in ACTN4 gene (Fig. 2). A strong linkage disequilibrium was observed between rs2112649 and rs3745859 (D' = 1, r2 = 0.5). In addition, a strong linkage disequilibrium was found between rs2287728 and rs899199, rs62120068, and rs12981131 (D' = 1, r2 = 0.93, 0.88, and 0.88, respectively) and rs899199 and rs62120068, rs12981131 sites (D' = 0.94 and 1, r2 = 0.88 and 0.94, respectively). The linkage analysis of rs62120068 and rs12981131 of ACTN4 gene revealed that the two loci had significant correlation (D' = 1, r2 = 1.00) (Fig. 2).

Linkage disequilibrium analyses of rs2112649, rs2287728, rs78078603, rs3745859, rs899199, rs62120068, rs12981131, and rs79072548 in ACTN4 gene.
Haplotype analysis of environmental factor correction
The p values for haplotypes ACCTCGG and ACCCCCG were less than 0.05, suggesting that there was significant difference between the case and control groups.
ACTN4 serum levels between the two groups
The serum ACTN4 concentration in the case group was markedly higher, averaging 544.7 ng/mL (range: 264.6-952.6 ng/mL), compared with 241.20 ng/mL (range: 110.75-542.35 ng/mL) in the control group. This difference was statistically significant (Z = 4.154, p < 0.01).
Discussion
Nikolopoulos et al. found that the ACTN4 is located on human chromosome 4 and encodes α-actinin-4 protein (Nikolopoulos et al., 2000). Mutations in the ACTN4 gene are a cause of autosomal dominant familial FSGS. It has been found that renal ACTN4 gene knockout can reduce the adhesion strength between foot process protein and basement membrane cells, leading to the destruction of filtration barrier (Kannan and Tang, 2015). Another study reported that heterozygous mutant mice with ACTN4 gene knockdown exhibited a significant improvement in proteinuria and glomerulosclerosis compared with homozygous mutant mice. This finding suggests a potential association between the ACTN4 gene and podocyte function (Read et al., 2014).
More and more studies have found that the mutation of ACTN4 gene is the primary genetic cause of delayed autosomal dominant familial FSGS (Welsh and Saleem, 2011; Wang et al., 2016). These patients usually begin receiving medical treatment in early adulthood (Welsh and Saleem, 2011). Feng et al. reported that a 14-year-old American child who was diagnosed with FSGS showed progression to end stage within 1 year of diagnosis (Feng et al., 2016). Through genetic testing, an Y265H variant with unknown clinical significance in ACTN4 was identified. This variant has not been seen previously in FSGS patients nor is it present in genetic databases (Feng et al., 2016). The authors suggested that ACTN4 with either Y265H or K255E (a known disease-causing mutation) increased the actin bundling activity of ACTN4 in vitro, was associated with the formation of intracellular aggregates, and increased podocyte contractile force (Feng et al., 2016). In this study, we performed additional cellular assays and found that the mechanism of action of Y265H mutation of ACTN4 gene is similar to that of K255E mutation of ACTN4 gene, which can lead to the increase of podocyte contractions leading to the occurrence of kidney diseases. Choi et al. (Choi et al., 2008) detected a new ACTN4 mutation (p.Ser262Phe) in Korean children who developed FSGS in their childhood (3-4 years old) and rapidly progressed to ESRD, and their father was found to have a germline chimeric mutation. Kaplan JM, et al (Kaplan et al., 2000) detected K255E, T259I, and S262P mutations in ACTN4 in three unrelated U.S. familial FSGS, and these mutations were associated with renal disease.
It is also reported in China that mutations in exon 10 of ACTN4 gene 947T > C (p.Leu316Pro) can cause familial FSGS, whereas mutations in promoter regions 1-34C > T, 1-590delA and (1-1044delT)+(1-797T > C)+(1-769A > G) can cause primary FSGS (Dai et al., 2010). Sun M et al. (Sun et al., 2014) conducted a study on 10 patients with kidney involvement in 50 members of a Chinese family with familial FSGS and found that ACTN4 gene had 184T > A (S62T) mutation, suggesting that this mutation may be one of the pathogenesis genes of familial FSGS. Similarly, Liu CY, et al. (Liu et al., 2017) also detected a heterozygous mutation of 184T > A (p.Ser62Thr) in exon 2 of ACTN4 gene in 78 Chinese patients with primary FSGS. However, this mutation was not detected in this patient’s parents or the 68 healthy controls, and no new pathogenic mutation was detected in other FSGS patients (Liu et al., 2017).
The above studies suggest that ACTN4 mutation is associated with the occurrence of nephrotic syndrome in different ethnic groups and different regions. In this study, c.1516G > A (p.G506S), c.1442 + 10G > A, c.1649A > G (p.D550G), c.2191-4G > A, and c.2315C > T (p.A772V) heterozygous mutations were detected only in the case group and were not present in 105 healthy controls. Data screening and biological information analysis showed that the above mutations can lead to abnormal expression and function of related proteins. For the novel missense variants p.G506S and p.D550G in the ACTN4 gene, and the variant p.A772V that is present in the gnomAD database and is listed as uncertain in ClinVar, these should be considered variants of uncertain significance until further functional analysis can be performed. The presence of c.2191-4G>A in 15 heterozygotes within an apparently unaffected cohort raises questions about its potential pathogenicity, especially considering the dominant inheritance pattern of ACTN4 associated disease. The presence of c.1442 + 10G > A in gnomAD and its absence in ClinVar leaves its pathogenicity uncertain. Considering that they are related to the onset of PNS in Zhuang children in Guangxi, China, they are pathogenic mutations. The c.162 + 6C > T, c.572 + 11G > A, and c.2191-5C > T heterozygous polymorphisms were detected in both case and control groups. The above polymorphisms detected in the control group are considered to be the mutation of ACTN4 gene susceptible to PNS in children of Zhuang nationality in Guangxi, China, but it has not yet occurred. In the later stage, children with related polymorphisms in the control group will be followed up regularly. The heterozygous mutation of c.162 + 9C > T, c.1691A > G (p.E564G), and c.1901A > C (p.D634A) was detected only in the control group. The above polymorphisms can lead to abnormal expression and function of related proteins, which may be susceptible mutations of PNS in Guangxi Zhuang children in China but have not yet occurred or may be pathogenic mutations of other diseases in these children, such as various tumor related diseases.
Follow-up observation will also be carried out on the above children. No heterozygous polymorphisms of c.1649A > G (p.D550G), c.1691A > G (p.E564G), c.1901A > C (p.D634A), and c.162 + 9C > T were found in the human mutation databases (dbSNP, 1000 Genomes, ESP6500, ExAC03, ExAC03_EAS, gnomAD, Hrcr1, Kaviar_20150923, GENESKYDB_Freq), and all were new ACTN4 mutations. Among them, heterozygous mutation of c.1649A > G (p.D550G) was only detected in case group, whereas c.1691A > G (p.E564G), c.1901A > C (p.D634A), and c.162 + 9C > T heterozygous mutation is not detected in the control group. One patient in the case group had heterozygous mutation of c.1649A > G (p.D550G) and c.2315C > T (p.A772V). All the above mutations were pathogenic by data screening and biological information analysis. These mutations can lead to abnormal expression and function of related proteins. However, due to the limited number of the cases, it is impossible to prove whether the two polymorphisms are linked. The correlation between the two gene polymorphisms and the incidence of PNS in Guangxi Zhuang children needs further research.
In summary, ACTN4 gene is a candidate gene for children with PNS. The mutation of this gene is closely related to the onset of PNS in children of Zhuang nationality in Guangxi, 3China. These mutations could facilitate diagnosis and aid in predicting the outcomes of clinical therapies. Moreover, it could possibly serve as a novel therapeutic target.
Authors’ Contributions
S.C., D.W., Y.H., and Z.H. mainly participated in literature search, study design, writing and critical revision; L.L., J.Y., L.W., Y.L., J Zhang, J Zhao, mainly participated in data collection, data analysis and data interpretation. All authors read and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.
Footnotes
Funding Information
This work was supported by Talents Training Program of Pudong Hospital affiliated to Fudan University (Project no. YJRCJJ201905), and Research and discussion on the pathogenesis of global growth retardation and sand table therapy in the treatment of children with global growth delay (Project no. YJRCJJ201912).