
Abstract
Background:
Osteosarcoma (OS), the most common primary malignant bone tumor, occurs mostly in the pediatric and adolescent (P/A) population where it has been subject to intense study whereas OS arising in the older-aged adult population has undergone less scrutiny.
Materials and Methods:
In this study, we assess the molecular aberrations detected in eight older adult patients (>59 years of age) with OS of bone by whole-exome sequencing (WES) on formalin-fixed, paraffin-embedded tissue and quantified the contributions of endogenous and exogenous mutational processes to tumor mutational burden and to tumorigenesis through computational analysis.
Results:
We identified 86 clinically significant somatic mutations. TP53 mutations occurred in OSs of three patients and one patient harbored a pathogenic germline mutation of TP53. Loss-of-heterozygosity of DNA-damage repair genes occurred in all six tumors evaluated. Computational analysis of single nucleotide variants within each tumor detected eight distinct mutagenic processes of which age-associated mutational processes, thiopurine chemotherapy, and defective homologous DNA recombination repair contributed the most to both tumor mutation burden and tumor pathogenesis.
Conclusion:
The genomic landscape of our older OS patients deciphered by WES is extremely diverse with only 15% of mutated somatic genes uncovered in our study previously described in P/A-enriched OS studies. Endogenous age-related mutagenic processes, defective DNA homologous recombination repair, and exogenous effects of chemotherapy are mainly responsible for pathogenic mutations in OS occurring in our cohort.
Introduction
Osteosarcoma (OS) of bone accounts for nearly 20% of all bone tumors (Fletcher et al., 2013) and is the most common primary malignant bone tumor. OS exhibits a bimodal age distribution with most cases arising in the pediatric population. A second peak occurs among older-aged adults (>65 years of age) (Cole et al., 2022; Mirabello et al., 2009; Negri et al., 2019; Savage and Mirabello, 2011; Rickel et al., 2017; Xu et al., 2019). Histologically, OS is defined by the presence of malignant mesenchymal cells directly producing osteoid or woven bone. The World Health Organization recognizes high-grade and low-grade intramedullary OS variants and three subtypes arising in association with the bone surface (Fletcher et al., 2013). High-grade intramedullary OS is further subtyped based on the principal stromal component which defines conventional osteoblastic, chondroblastic, or fibroblastic variants; distinct cytomorphologic features (e.g., small-cell, epithelioid, clear cell, and giant cell-rich variants) or unique architectural patterns (e.g., telangiectatic OS). Lastly, the dedifferentiated OS is a high-grade sarcoma that arises from low-grade OS (Fletcher et al., 2013).
Pediatric and adolescent (P/A) OS and OS arising in older adults display significant clinicopathologic and etiologic differences (Mirabello et al., 2009). OS in older adult patients is typically more aggressive, presenting mostly as a pure lytic lesion, and tends to involve the axial skeleton and craniofacial bones rather than the metaphysis of long bones, where rapid cellular proliferation within the prepubertal skeleton is responsible for bone lengthening (Huvos, 1986; Mirabello et al., 2009; Naka et al., 1995; Okada et al., 2004; Tsuda et al., 2018). The morphology of older adult OS often reflects its more aggressive clinical course being composed of pleomorphic spindle cells reminiscent of soft-tissue undifferentiated pleomorphic sarcoma but with scant amounts of neoplastic bone (Huvos, 1986; Naka et al., 1995). In further contrast to P/A OS, tumors arising in older adult patients are often associated with pre-existing bone conditions (Huvos, 1986), most commonly Paget’s disease of bone or prior bone exposure to radiation (Huvos et al., 1983). These contrasting clinicopathologic attributes raise speculation that differences in OS molecular pathogenesis exist between these two patient populations.
Molecular studies focusing mainly on P/A OS have shown marked genomic instability, manifesting as diverse clonal and subclonal gene alterations, frequent involvement of chromosomal mutagenic processes, chromothripsis and kataegis, and the highest rate of somatic mutations among childhood tumors (Rickel et al., 2017). Alterations in numerous candidate driver genes affecting diverse pathways including TP53, RB1, CDKN2A, PTEN, DLG2, ATRX, MYC, and RUNX have been detected by whole-genome or exome genomic analysis of mainly P/A OS (Berman et al., 2008; Chen et al., 2014; Freeman et al., 2008; Hou et al., 2020; Kovac et al., 2015; Mohseny et al., 2010; Rickel et al., 2017; Smida et al., 2017). In addition, a unique TP53 intron 1 rearrangement has been found in nearly 50% of pediatric examples of OS, but not in adult OS (Chen et al., 2014; Marrano et al., 2018). Studies assessing germline mutations in pediatric OS have identified mutations in TP53 (Li-Fraumeni Syndrome), RB1 (Hereditary Retinoblastoma), REQL4 (Rothmund-Thomson Syndrome), WRN (Werner Syndrome), BLM (Bloom’s Syndrome), and in ribosomal protein genes (Diamond-Blackfan Anemia) (Savage and Mirabello, 2011) that lead to well-recognized familial hereditary cancer syndromes.
In this study, we collected clinicopathologic data and identified tumoral and normal tissues from eight older adult OS cases and from four control undifferentiated pleomorphic sarcoma of bone (UPSb) cases from similarly aged patients. We performed whole-exome sequencing (WES) and loss-of-heterozygosity (LOH) analysis to characterize the genomic landscape of OS in older adult patients where the rate of diagnosis is increasing, especially in patients over 60 years of age (Nishida et al., 2009) and molecular data reported in this OS population is scant (Kawaguchi et al., 2002). In our adult OS cohort, we identified genes and genetic characteristics likely to play oncogenic roles, compared genomic burdens attributable to tumor mutational processes, and assessed contributions of mutational processes to tumorigenesis.
Materials and Methods
Patient sample accrual
After Institutional Review Board approval and in accordance with ethics committee criteria of the Yale School of Medicine, we searched in our electronic medical records system for archived pathology cases from 1980 to 2019 coded as “osteosarcoma of bone” in patients 59 years or older. Eight cases formed the basis of our study (Table 1). We also retrieved four high-grade undifferentiated sarcomas coded as “malignant fibrous histiocytoma of bone,” “undifferentiated sarcoma of bone,” or “fibrosarcoma of bone” in similarly aged patients as a comparison group. To confirm the diagnosis and select blocks with representative tumor and corresponding normal tissue, slides from all the tissue blocks of the OS and UPSb study cases were reviewed by two pathologists with interest in bone and soft tissue pathology (W.B.L. and H.K.) (Fig. 1).

Representative histological images from an elderly adult osteosarcoma and pleomorphic undifferentiated sarcoma of bone case.
Clinicopathologic Characteristics of Adult Osteosarcoma Patients (8 Patients)
F, female; M, male.
Whole-Exome sequencing and loss-of-heterozygosity analysis
We performed exome sequencing on formalin-fixed paraffin-embedded blocks from the eight OS and four UPSb cases culled from the surgical pathology archives of Yale-New Haven Hospital. Six OS and three UPSb cases with matched normal tissue were evaluated for germline mutations and LOH. Tumor DNA was extracted from formalin-fixed paraffin-embedded tissues using the Biostic FFPE Tissue DNA Isolation kit (MO BIO Laboratories, 12250-50). DNA (2-3 μg/subject) was sent to the Yale Center for Genome Analysis for WES. We utilized a standardized commercial tumor-profiling IDT xGen human Exome V2 panel test (528 genes) utilized by our cancer center for genetic testing in solid tumors. DNA was sheared to a mean fragment length of 220 bp using focused acoustic energy (Covaris E220, Woburn, MA). Fragments were then blunt-ended and phosphorylated using T4 DNA polymerase and T4 polynucleotide kinase. Custom adapters were ligated to each fragment using T4 DNA ligase before DNA fragments were PCR amplified. The amplified DNA was then heat-denatured and mixed with biotinylated DNA probes (IDT xGen Exome Panel, Coralville, IA). Hybridizations were performed at 65 Centigrade for 16 h. Captured fragments were PCR amplified, then purified with AMPure XP beads to generate each a DNA library for each subject. 2 × 100 bp paired ends of these libraries were sequenced using the Illumina Novae 6000 S4 platform. Burrows-Wheeler Aligner was used to map sequence reads to the genome. The Yale Exome Pipeline was used to call exome-wide variants and Annovar and Variant Effect Predictor were used for variant annotation. Average coverage was at least 250×. Variants detected by a minimum read depth less than 20 or by less than 5 high-quality reads were filtered out. The clinical significance of the genetic mutations was analyzed utilizing multiple databases including COSMIC, ClinVar, SIFT, PolyPhen, and CADD.
Cancer effect size and tumor mutational burden analysis
We assessed the contribution of each mutation to cancer cell survival and proliferation (Cannataro et al., 2018; Cannataro et al., 2022) using cancer effect size R software package (v2.3.4) (Blokzijl et al., 2018; Mandell et al., 2023). Cancer effect size accounts for underlying rates of mutation at each site in each gene and quantifies for each variant its impact on the malignant potential of the neoplastic cell (Cannataro et al., 2018). We also analyzed the influence that endogenous and exogenous mutagenic processes have on formation of oncogenic driver mutations. Applying trinucleotide mutational signature deconvolution to the data set defined eight distinct mutagenic processes: Clock-like deamination with age (COSMIC Single-Base Substitution signature SBS1), Apolipoprotein-B mRNA-editing enzyme catalytic polypeptide (APOBEC; SBS2, SBS13), Defective DNA homologous recombination repair (SBS3), Unknown, clock-like (SBS5), Defective DNA-base exclusion owing to NTHL1 mutation (SBS30), Unknown chemotherapy treatment (SBS86), Thiopurine chemotherapy treatment (SBS87), and Nonactionable or unknown signature (SBS8, SBS17a) (Fig. 2).

Mutational tumor burden weights and cancer effect associated with the mutational signatures in osteosarcomas of elderly adult patients (8 patients). (
Results
Clinicopathological characteristics
The clinicopathological characteristics of our eight-patient cohort are summarized in Table 1. The median age of our cohort was 76 (range: 59-84) years. Six of the eight OS patients presented with the tumor in the axial skeleton or craniofacial bones and all but one tumor was a primary sarcoma. Three patients developed lung metastases. Significant risk factors for the development of OS include prior radiation treatment for an unrelated malignancy in the same area as the OS (n = 2) and Paget’s disease of bone (n = 3). Three patients had documented evidence of neoadjuvant chemotherapy for their OS.
WES identifies recurrent and clinically somatic significant mutations in OS
We detected many pathogenic/likely pathogenic somatic mutations and variants of undetermined significance on WES in our OS cohort (Table 1). Ten mutations were recurrent in three of the eight patients (38%), all of whom harbored TP53 mutations. Most cases demonstrated at least two pathogenic/likely pathogenic significant mutations with potential clinical significance (median: 2). To identify potential OS oncogenic driver genes, we focused on somatic mutations detected in candidate genes reported primarily in P/A OS patients in the recent literature and found 12 matching genes in our cohort (Table 2; Supplementary Table S1) (Bousquet et al., 2016; Hou et al., 2020; Perry et al., 2014; Rickel et al., 2017). Pathogenic somatic substitutions were found in two BRCA-related genes, ATR and RAD51 (n = 1 each). In addition, substitutions in osteogenesis-related BMP genes (n = 1) and APC2 (n = 1) were detected. Substitutions in MYO7B and PCDH15, previously described in UPSb (Ali et al., 2019), were found in one case each.
Driver Mutations Reported in Pediatric/Adolescent OS-Enriched Studies and Detected in Our Adult Osteosarcoma of Bone (OS) Patients (8 Patients)
LOH in DNA-damage repair genes
We observed substantial LOH in DNA-damage repair (DDR) genes including those in the homologous recombination DNA repair and BRCA pathways in all six OS cases with corresponding normal tissue analyzed (Table 3). LOH was found in BAP1 (n = 5), BRCA1/2 (n = 4, each), RAD50 (n = 4), CHEK2 (n = 4), CHEK1 (n = 3), RAD51 (n = 3), PALB2 (n = 3), ATM (n = 2), ATR (n = 1), and in POLE and POLD1 (n = 2, each), genes encoding the catalytic units of delta and epsilon DNA polymerase.
Somatic Mutations (8 Patients) and LOH (6 Patients) in Important DNA-Damage Repair (DNA Homologous Recombination Repair and BRCA-Related) Genes in Adult Osteosarcoma Patients
LOH, loss-of-heterozygosity.
Exome sequencing identifies a germline mutation
We identified one tumor with a pathogenic germline nonsense mutation in TP53, which has previously been described in hereditary cancer-predisposing syndrome.
Mutational processes and TMB and cancer effect size analysis
Mutational processes contributing the most to tumor mutational burden (TMB) were the nonspecific (unknown, clock-like) aging process, deamination with age, thiopurine chemotherapy, and defective DNA homologous recombination repair (Fig. 2). Processes resulting in mutations with the greatest cumulative effect on tumorigenesis (cancer effect) were the nonspecific (unknown, clock-like) aging process, thiopurine chemotherapy, and defective DNA homologous recombination repair (Fig. 2). Deamination with age contributed proportionally more to exome TMB than it did to cancer effect, whereas the nonspecific (unknown, clock-like) aging process and defective DNA homologous recombination repair contributed proportionally more to cancer effect than to TMB. Thiopurine chemotherapy contributed nearly equally to TMB and cancer effect.
Comparison to UPSb
The four UPSb patients consisted of three males and one female ranging from 59 to 94 years of age (Fig. 3). Three sarcomas arose in the femur and one in the tibia. The histology of normal bone tissue from the femoral mass in the 94-year-old male also showed features of Paget’s disease. A prior history of chronic osteomyelitis involving the tibia was documented for the 87-year-old male. We detected many somatic mutations in this cohort, of which some were recurrent and 26 (37%) were also found in our OS cohort (Fig. 3). Pathogenic mutations in TP53 were found in three cases (3/4). Somatic mutation in the DDR-related genes ATM was identified in one tumor. In the three UPSb cases tested, LOH was found in DDR-related genes, ATM, PALB1, POLE, CHEK2, BAP1, BRCA2, RAD51 (n = 2, each), and CHEK1, ATR, and POLD1 (n = 1, each). Aging processes, defective DNA homologous recombination repair, and thiopurine chemotherapy were mutagenic processes that significantly impacted both TMB and cancer effect (Fig. 3).
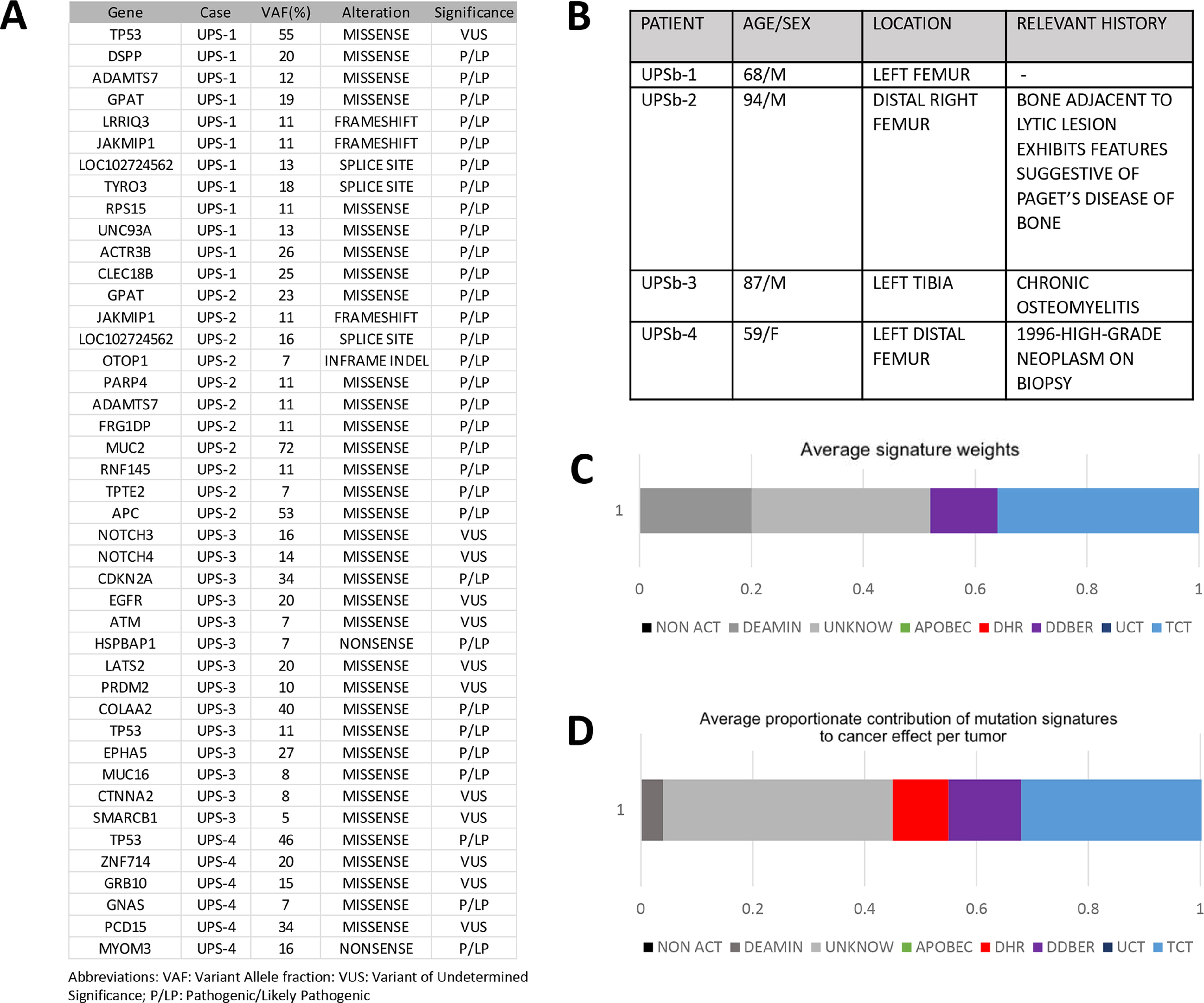
Mutational tumor burden weights and cancer effect associated with mutational signatures in undifferentiated pleomorphic sarcoma of bone patients (4 patients) (
Discussion
In the current study, we assessed somatic mutations and LOH of DDR-related genes present in a cohort of eight older-aged adult OS patients by WES. We compared our results with molecular data previously published in five large P/A-enriched studies of OS and with WES results obtained from four examples of UPSb from similarly aged patients culled from our files. Lastly, we utilized computational analysis to assess the TMB and possible tumor etiology based on molecular signature (cancer effect size) in our OS and UPSb cohorts.
Assessment of genetic mutations in our cohort revealed 86 somatic mutations, of which 10 were recurrent affecting diverse signaling pathways, including cell cycle/apoptosis pathways, the DDR pathway, and histone and chromatin modifier pathways. Numerous additional passenger mutations and mutations of undetermined clinical/oncological significance were also identified. TP53 mutations were the most commonly recurrent clinically significant somatic mutation observed in our cohort (3/8 tumors; 38%) (Supplementary Table S1). We and others believe that TP53 alterations are likely early events in sarcoma pathogenesis and facilitate genomic instability (Kovac et al., 2015) as evidenced by the presence of at least one other significant somatic mutation in our cases with TP53 mutation. The somatic mutations detected in our OS cohort are similarly diverse as those previously reported in studies focusing mostly on pediatric and adolescent OS (Bousquet et al., 2016; Chen et al., 2014; Nacev et al., 2022), but we found only 12 genes in our study that were previously reported in five select series (Bousquet et al., 2016; Chen et al., 2014; Hou et al., 2020; Kovac et al., 2015; Perry et al., 2014). Moreover, of the purported oncogenic driver genes reported in P/A OS, ATRX, and DLG2 mutations were not detected in our study, and we found only three cases with TP53 mutations and one case each with either a RB1 or PTEN mutation (Bousquet et al., 2016; Chen et al., 2014; Hou et al., 2020; Kovac et al., 2015; Rickel et al., 2017; Perry et al., 2014). Our results add further evidence that somatic mutations in older adult OS like P/A OS are diverse, primarily nonclonal, and result from events leading to genomic instability and intratumoral heterogeneity (Rickel et al., 2017).
In addition, unlike P/A OS where germline mutations are more frequently encountered, we found only one pathogenic germline mutation—a nonsense germline mutation in TP53 previously described in Li-Fraumeni hereditary cancer-predisposing syndrome.
In keeping with previous studies with emphasis on OS (Kovac et al., 2015; Nacev et al., 2022), we identified LOH in BRCA-related genes in six tumors with corresponding normal tissue. The high incidence of DDR pathway gene and TP53 alterations could potentially lead to significant impairment of the homologous recombination DNA repair pathway, which is targetable with PARP inhibitors. Although these results are interesting, the lack of gene expression data precludes definitive determination of whether these LOH are “by-standers” of genomic instability or participate in sarcoma pathogenesis (Kuijjer et al., 2012).
The mechanisms responsible for such profound genomic instability observed in our cohort were not investigated, but two processes implicated at least in part for genomic instability witnessed in OS have been described in studies heavily enriched with P/A patients. Localized chromosomal base-pair hypermutations (kataegis) commonly encountered in TP53 and ATRX genes have been reported in 50-85% of cases (Chen et al., 2014; Cortés-Ciriano et al., 2020) and chromothripsis, a process of chromosomal fragmentation resulting in rearrangements, gene loss, and copy number gains from formation of circular extrachromosomal amplified DNA, has been documented in 50-77% of tumors (Behjati et al., 2017; Chen et al., 2014). To date, these mechanisms have not been studied in a pure older adult OS patient population.
We also compared molecular findings from a small number of UPSb cases with our cohort of OS as both lesions often demonstrate overlapping clinical and histological features. We identified 37% of the somatic mutations found in our UPSb cases also in our OS cohort as well as LOH in DDR genes in the three UPSb tumors evaluated. A recent study of UPSb detected frequent mutations in TP53 and in chromatin-remodeling genes DOT1L, H3F3A, and ATRX, as well as overexpression of the osteoblast/osteocyte-derived phosphatonin, FGF23. Similar to our OS cases, TP53 was the most common clinically significant mutated gene occurring in three of four UPSb cases, but no mutations in DOT1L, H3F3A, or ATRX were found (Ali et al., 2019). Taken together, our comparison suggests a close histogenetic relationship between UPSb and OS occurring in older-aged individuals.
We evaluated the impact that various etiologic processes defined by trinucleotide mutational signatures had not only on the mutational burden but also on sarcoma pathogenesis (cancer effect size) (Cannataro et al., 2022). Our analysis demonstrated that the mutational processes related to aging (either due to deamination with age or to unknown, clock-like aging process), thiopurine chemotherapy, and defective DNA homologous recombination repair contributed to both TMB and cancer effect, with the largest contributor to both TMB and cancer effect coming from the nonspecific (unknown, clock-like) aging process. The nonspecific (unknown, clock-like) aging process and defective DNA homologous recombination repair contributed more to cancer effect than to TMB. These results differ from data of most epithelial neoplasms (Cannataro et al., 2022). However, as in brain tumors and gastrointestinal epithelial tumors, signatures related to aging processes (#1 and #5) were a substantial contributor to TMB and cancer effect in our OS population (Cannataro et al., 2022). The paired contributions of aging processes and defects in DNA homologous recombination repair to tumorigenesis in our cohort raise speculation that the latter may also be an age-dependent phenomenon. The important contribution of age-related processes to the development of OS in our elderly cohort could be explained in part by age-dependent telomere shortening, which plays a salient role in sarcoma pathogenesis (Ballinger et al., 2023). Thiopurine chemotherapy contributed significantly to both the TMB and cancer effect, although only three patients had documented evidence of receiving chemotherapy. As the full medical record was not available for most patients in this cohort because either the patients were external consultations or because the electronic medical record was not yet implemented; information regarding exposure to chemotherapy in all cases could not be assessed. Moreover, the nonspecific (unknown, clock-like) aging process, thiopurine deficiency, and defective homologous recombination repair were also the three largest contributors to cancer effect in the UPSb cases we studied, indicating a similar etiology for both UPSb and OS in older-aged patients.
Our small study demonstrates that elderly OS and UPSb patients harbor a diverse array of genetic alterations including somatic mutations and LOH like P/A OS, but except for TP53 share only a small number of mutated somatic genes with the latter suggesting a different pathogenesis. DDR gene alterations, mostly LOH, were common in our OS and UPSb cases. Our computational analysis indicates that mutagenesis in older adult OS and UPSb and ultimately pathogenesis of these sarcomas appear driven by endogenous factors related to aging, defects in DDR pathways, and in select cases, remote exposure to chemotherapeutic agents, and that OS and UPSb in older adult individuals share a close histogenesis.
Footnotes
Acknowledgments
The authors thank Yale Center for Genome Analysis for their technical help in the WES.
Authors’ Contributions
H.K. and W.L.: Designed the study, analyzed the data, and wrote the article. J.C. and J.T.: Actively participated in reviewing the paper and played a significant role in editing the article. J.T. and C.T.: Performed the cancer effect/TMB studies. All authors have provided substantial contributions to this paper, endorsed the final version for submission, and committed to taking responsibility for all aspects of the work.
Compliance with Ethical Standards
The writing of this work was in full compliance with ethical standards.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.