
Abstract
Slow transit constipation (STC) is a complication of depression that can negatively impact patient prognosis and quality of life. Nonetheless, the pathogenesis of STC is unclear. In this work, colon tissues from STC and non-STC patients were utilized to determine transcriptome expression patterns (messenger ribonucleic acids [mRNAs], Long noncoding RNAs [lncRNAs], and Circular RNAs [circRNAs]) via high-throughput sequencing. We found that 4430 mRNAs, 984 lncRNAs, and 2152 circRNAs exhibited substantial variations in expression patterns in the colon tissues of STC and non-STC patients. Next, we constructed a protein-protein interaction network and identified three significant elements, namely, POLR2B, SRSF1, and SUMO1, which attracted our interest. Utilizing the data of 6 upregulated circRNAs and 10 downregulated circRNAs, we created a competing endogenous RNA network. Subsequently, we found that hsa_circ_0000994 and hsa_circ_0008699 were significantly enriched in the upregulated and downregulated networks, respectively. The coexpression network analysis suggested that circRNAs and lncRNAs might exert control over mRNAs by influencing the neural functions of STC. According to the results of the integrated circRNA-miRNA-mRNA network, circRNA-regulated mRNAs were linked to both the transforming growth factor-β (TGF-β) and Notch signaling pathways. Our findings could provide new perspectives for identifying potential prognostic markers in STC. Targeting SUMO1 may present a promising approach to address colonic motility disorders in STC therapy.
Introduction
Slow transit constipation (STC) is characterized by reduced intestinal motility and prolonged colonic transit time and is characterized by delayed colonic transit, a lack of urge to defecate, and a considerable decrease in stool frequency (Bharucha and Wald, 2019). STC is becoming more common as the pace of modern life accelerates, living standards rise, diets change, and the social population ages (Bharucha and Lacy, 2020). Nyrop et al. studied constipation and medical expenses and discovered that the severity of a patient’s condition is proportional to medical expenses. Moreover, constipation has become one of the most common disorders harming modern people’s quality of life. Constipation is a significant social public health issue because of the widespread concern it has sparked in society due to its high occurrence rate (Nyrop et al., 2007). Most of the symptoms of chronic constipation can be alleviated by changing one’s lifestyle, adjusting one’s diet structure, or administering pharmacological adjuvant therapy; however, medical medication therapy has no effect on intractable STC (Daniali et al., 2020). Long-term constipation is also a major risk factor for perianal disorders such as hemorrhoids, anal fissure, and rectal prolapse, with apparent consequences. Currently, numerous investigations have revealed that STC has numerous pathogenic components and complex mechanisms. The following topics are covered in current research on the etiology and mechanism of STC: intestinal flora disorders, hormonal anomalies, psychosocial factors, autoimmunity, and other factors. Gaining a comprehensive understanding of pathophysiology from a single viewpoint is challenging, and the clinical treatment outcomes are unsatisfactory (Prichard and Bharucha, 2018). In the field of digestion, explaining the pathophysiology of STC is difficult. This highlights the critical need for additional biomarkers to enable more accurate diagnosis and treatment predictions for STC.
Advances in science and technology have revealed that mRNAs, lncRNAs, and circRNAs play regulatory roles in cellular functions in animals (Boivin et al., 2020). Numerous lncRNAs are involved in nuclear transport, transcriptional activation, and interference, as well as chromosomal and genomic modifications. This has led to increased interest among researchers in understanding the influence of lncRNAs on human biology. Initially, lncRNAs were thought to be secondary products of RNA polymerase II transcription and were not expected to be synthesized into proteins (Dahariya et al., 2019). Many lncRNAs have been detected in colon tissues, and it was expected that they would play a substantial role in the development of STC in rats (Yan et al., 2021). CircRNAs are extensively dispersed throughout mammalian cells and play various regulatory roles in controlling gene expression at both the transcriptional and posttranscriptional levels (Nowak et al., 2022). However, miRNAs are an alternative type of noncoding RNA that has been associated with the initiation and progression of STC. Research conducted by Gong et al. demonstrated that miR-30b-5p can influence the progression of STC by suppressing the activity of PI3K/Akt/mTOR (Gong et al., 2022). Both circRNAs and lncRNAs serve as inhibitors of miRNAs, functioning as molecular sponges to modulate mRNAs (Ma et al., 2023; Militello et al., 2017). However, the functions of circRNAs, lncRNAs, and mRNAs in STC are unknown.
The screening of genetic variation at the genome level using RNA sequencing and bioinformatics analysis has been popular in recent years. This approach can reveal potential molecular mechanisms underlying the emergence of diseases and offer workable concepts for experimental investigation. In this study, bioinformatics techniques were used to evaluate the expression of RNA in colon tissue from STC patients to explore the gene expression and biological metabolic processes of STC and to discover potential therapeutic indicators.
Materials and Methods
Patients and tissue samples
This study included a total of 10 participants: 5 individuals diagnosed with STC who underwent subtotal colectomy and 5 control subjects who underwent radical surgery for colon cancer. We chose patients with STC in strict accordance with international and national guidelines. The inclusion criteria were as follows (Deng et al., 2023; Kamm et al., 1988; Knowles et al., 1999; Wang et al., 2023): (1) met the Rome IV STC diagnostic criteria; (2) had severe constipation symptoms, a defecation frequency less than twice per week, no urge to defecate or no obvious desire to defecate, a high incidence of abdominal distension, and laxatives must be used to maintain defecation or laxatives cannot maintain defecation; (3) constipation sensations had been present for more than 5 years, substantially impairing quality of life, and medical treatment was ineffective for more than 2 years, with a strong desire for surgery; (4) colorectal organic lesions were excluded; (5) there was no evident stomach or intestinal motility impairment based on three colonic transit tests; (6) a Hamilton Anxiety Scale <14 points and no visible mental symptoms such as depression; and (7) the constipation kind of irritable bowel syndrome was excluded.
Before the operation, all patients underwent a gastrointestinal transit test. The running track of the opaque marker was used to locate the intestinal segment associated with colonic transit dysfunction, and the normal intestinal segment of colon cancer patients was chosen as the control. Each sample was collected from the colon tissue within 3 min after surgery, divided into 1 cm3 samples, and then cryopreserved in liquid nitrogen at −80°C. The thorough records of all the patients involved in the study are presented in Table 1. Moreover, two pathologists confirmed that the constipation samples exhibited pathological alterations associated with delayed transit constipation, such as reduced intestinal nerve cells and decreased interstitial cells of Cajal (ICC) cells (Fig. 1). Colon specimens were collected from STC patients who underwent surgery at the Department of Colorectal Surgery, Third Affiliated Hospital of Henan University of Chinese Medicine, from September 2017 to July 2018. Tumor-free control samples were obtained from colon cancer patients who underwent surgery at the Department of Anorectal Surgery, Suzhou TCM Hospital Affiliated with Nanjing University of Chinese Medicine, from September 2017 to July 2018.

Preoperative barium-strip examination
Basic Characteristic of Patients with STC and Control Individuals
Data are presented as mean ± SD or number.
STC, slow transit constipation.
Each patient provided informed written consent, and the study received approval from the Ethics Committee of Suzhou TCM Hospital Affiliated with Nanjing University of Chinese Medicine (ethical application ref: 201703003) and the Third Affiliated Hospital of Henan University of Chinese Medicine. Colon tissue samples were collected from patients with STC and colon cancer patients in accordance with the principles of the Helsinki Declaration. The research was formally registered on the Chinese Clinical Trial Registry with study ID ChiCTR-BON-17012324.
RNA extraction, library preparation, and sequencing
We acquired total RNA from the 10 samples by employing TRIzol reagent from Invitrogen (San Francisco, CA, USA). We utilized a total of 3 grams of RNA as the input material for RNA sample preparation. After determining the quality and concentration of the RNA and removing ribosomal RNA, RNA-Seq libraries were generated following the manufacturer’s guidelines for the NEBNext® UltraTM Directional RNA Library Prep Kit for Illumina® (NEB, USA). Finally, we performed sequencing on all the libraries using the Illumina HiSeq 4000 system, which yielded 150-base pair paired-end sequencing reads.
Quality control, alignment, and quantification of RNA-seq data
Fastq unprocessed data were cleaned using in-house Perl scripts. Reads containing adapters or of low quality were deleted, and the Q20, Q30, and GC content of the clean data were calculated to evaluate the quality of the sequencing. HISAT2 was used to align paired clean reads with the rat reference genome (rn6) (v2.1.0). StringTie (v1.3.3) was used to construct the transcripts for each sample based on the aligned reads using an annotated transcript file from the ENSEMBL database. The coding potential of undocumented transcripts (new transcripts) was estimated using the Coding-Non-Coding Index (v2), Pfam Scan (v1.3), and Coding Potential Assessment Tool (v1.2.4). When three tools were concurrently reported with or without coding capacity, previously undiscovered mRNAs or lncRNAs were identified. StringTie was subsequently employed to determine the fragments per kilobase of transcript per million mapped fragments (FPKMs) of both mRNAs and lncRNAs (v1.3.3).
We employed CIRCexplorer (v2.2.3) for the detection of circular junctions and spliced sequences, leveraging the fusion junctions acquired from TopHat2 to establish the presence of circRNAs. We classified candidates with a minimum of 2 junction reads as genuine instances of circRNAs and used transcripts per million to evaluate circRNA expression levels.
Discovery of variably expressed transcripts
We utilized DESeq2 to identify differentially expressed mRNAs (DE mRNAs) and lncRNAs (DE lncRNAs) between the STC and control groups, and we employed the limma tool in R to pinpoint dissimilarly expressed circRNAs. The statistical significance thresholds for this analysis were set at a p value of 0.05 and an absolute log2-fold change of 1.
Protein-protein interaction network construction
We used the STRING database (v 11.0) to identify possible connections between proteins translated from the top 300 DE mRNAs (based on |log2-fold change|), with a confidence score of 0.7 chosen. We constructed the protein-protein interaction (PPI) network graphically by using Cytoscape software (v3.8.0, http://www.cytoscape.org/).
Investigation of genes targeted and influenced by lncRNAs
The primary purpose of lncRNAs is to influence target genes either locally (cis) or distantly (trans). Cis-action can be explained by the fact that lncRNAs might control nearby protein-coding genes. As a result, for the lncRNAs, DE mRNAs located 100 kb before or after the DE lncRNAs were chosen as targets. A lncRNA-mRNA coexpression network was constructed through the mutual regulatory interactions between DE lncRNAs and DE mRNAs, which were subsequently created for trans-acting genes. We computed the Pearson correlation coefficient (PCC) by utilizing log2 (FPKMs + 1), and the section with |PCC| 0.98 and a p value of 0.001 was chosen as noteworthy. Finally, the resulting network was visualized using Cytoscape software.
CircRNA-miRNA-mRNA network construction
We evaluated the circRNA-miRNA-mRNA interactions via miRanda (v3.3a) and selected upregulated and downregulated circRNAs/mRNAs to construct the competitive endogenous RNA (ceRNA) network. The network was visualized via Cytoscape software.
Functional enrichment analysis
DAVID (v6.8) and KOBAS (v3.0) software were used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses for the genes of the DE mRNAs of the host. Significant enrichment was determined by a p value of 0.05.
Chemicals, drugs, and reagents
Loperamide hydrochloride (Lot number: L4762-5G) was procured from Sigma Chemical Co., St Louis, MO, USA. Primary antibodies against SUMO1 (Lot number: ab 5316), c-Kit (Lot number: ab114992), POLR2B (Lot number: ab51362), and SRSF1 (Lot number: ab114675) were purchased from Abcam (Cambridge, UK). TRIzol (Lot number: 10296010CN) was provided by Invitrogen, USA. A reverse transcription kit was obtained from Takara, Japan. All other chemicals and reagents used in the experiment were analytical grade.
Animal model establishment
Each animal study protocol received evaluation and approval from the Institutional Animal Care and Use Committee (IACUC) in accordance with international standards. The number of animals utilized in each study was minimized to the necessary amount to meet study objectives, considering regulatory requirements, statistical power, and historical data availability. All experimental procedures described in this article were conducted with the utmost consideration for animal welfare. The reporting of animal studies adheres to the ARRIVE guidelines (Kilkenny et al., 2010). Male C57 mice were purchased from Hunan Slake Jingda Experimental Animal Co., Ltd. (Changsha, China) with certificate number SCXK (Xiang) 2019-0004. All mice were maintained in a controlled environment at 23°C with a 12-h dark/light cycle for 1 week and provided with free access to food and water. All mice used in our research were operated under a protocol approved by IACUC of Nanjing University of Chinese Medicine (Ethics approval L20250177; February 12, 2025). According to the research literature on relevant constipation mouse models (Li et al., 2023; Wen et al., 2023), 16 male C57 mice were required for the animal experiment, and the mice were randomly divided into two groups (8 mice in each group): control and STC. All mice were sacrificed on the 15th day, and after fasting for 12 h, the mice were anesthetized by intraperitoneal injection of 1% sodium pentobarbital. Colon tissues were collected, and the length and weight of the colon were recorded.
Fecal particle count and fecal water content
The fur color, mental state, and activity of mice were observed every day, and the body weight and food intake of mice were monitored. After the intervention, the mice were fasted but not watered for 16 h. The feces of each mouse within 6 h were collected the next day; the number of particles was recorded and then weighed and recorded as the wet weight of feces. The feces were placed in a constant temperature box and baked at 90°C for 3 h. After drying, they were weighed again and recorded as the dry weight of feces. The water content of feces was calculated, and the water content of feces (%) = (wet weight of feces − dry weight of feces)/wet weight of feces × 100%.
Measurement of intestinal transit function
10% activated carbon suspension was gavaged, and the samples were collected 30 min later. The entire small intestine was taken out, and the small intestine was peeled off without traction to make it flat; the distance from the mouse pyloric sphincter position to the end of carbon powder propulsion and the total length of the small intestine were measured and recorded with a ruler, and the small intestine propulsion rate was calculated. Small intestine propulsion rate (%) = carbon powder propulsion distance in the intestine/total length of the small intestine × 100%.
Histopathological observation of mouse colon tissue
The distal colon tissue of mice was taken and repeatedly rinsed with saline, fixed in 4% paraformaldehyde, dehydrated, transparent, waxed and embedded, and sliced after waxing. Hematoxylin-eosin (HE) staining was used to observe muscular thickness and colonic pathological changes, and semiquantitative analysis of colonic pathological changes was performed.
The overall morphological scoring of colonic tissue is as follows (Du et al., 2018): A score of 0 is assigned when the colonic tissue structure is clear and villi are neatly arranged; a score of 1 is given when the colonic tissue structure is vague but villi are neatly arranged; and a score of 2 is assigned when the colonic tissue structure is vague and villi are disorganized. For epithelial cell shedding, a score of 0 indicates no shedding of colonic epithelial cells; a score of 1 indicates shedding of colonic epithelial cells without forming sheets; and a score of 2 indicates shedding of colonic epithelial cells that form sheets. For inflammatory cell infiltration, a score of 0 is assigned when 0-5 inflammatory cells are observed under the microscope; a score of 1 is assigned for 6-10 inflammatory cells; and a score of 2 is assigned for more than 10 inflammatory cells observed. Alcian blue-periodic acid Schiff staining was used to observe the colon muscle layer, mucosal layer thickness, goblet cell number, mucus area in the mucosal layer, and other colon histological parameters. For each section, eight random images were captured at 400× magnification from different locations. The integrated optical density and the effective statistical area were measured using Image-pro-plus 6.0 software. The mean optical density was calculated, and subsequent analysis was performed using the average values obtained (Chen et al., 2024).
Determination of reverse transcription PCR
Total RNA was extracted from tissue by Trizol (Invitrogen, Carlsbad, CA, USA). After determining the purity by ultraviolet radiation, the concentration of total RNA was adjusted to the same level in each group. The reverse transcription product can be directly used for real-time PCR reaction for amplification. Primers were designed using Primer 5 software. PCR primers were synthesized by Qingke Biotechnology (Beijing) Co., Ltd. The sequences of each primer are shown in Table 2. Quantitative PCR amplification program: 95°C predenaturation for 10 min, enter the following cycle: 95°C denaturation for 15 s; 60°C annealing for 30 s, for a total of 40 cycles. glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal reference, and the expression level of the target gene was calculated by the relative quantification method. The relative expression amount was expressed as 2−ΔΔCt. The experiment was repeated three times for each sample.
Primers for Quantitative Real-time PCR (qPCR) Examination
Immunohistochemistry
Paraffin-embedded tissue sections were first rehydrated in xylene and then in graded ethanol solutions. For antigen retrieval, the sections were immersed in 0.01 M citrate buffer (pH 6.0) and heated to boiling using an electric or microwave oven. The heat was then turned off, and the sections were allowed to boil for 20 min. After cooling for 20 min, the sections were brought to room temperature. Subsequently, the sections were washed with 0.01 M PBS (pH 7.2-7.6). 1% periodic acid was added and incubated at room temperature for 10 min to inactivate endogenous enzymes and then rinsed with PBS. After blocking with normal goat serum, the sections were immunostained with primary antibodies c-Kit (1:300), POLR2B (1:200), SRSF1 (1:250), and SUMO1 (1:100) and incubated overnight at 4°C. Then, the sections were washed with PBS and incubated with secondary antibodies for 30 min at room temperature. Subsequently, the sections were washed with PBS and incubated in DAB kit for 1-5 min at room temperature. The sections were counterstained with hematoxylin, rinsed with distilled water, and blued with PBS. Dehydration and transparency: dehydration with different levels of alcohol (60-100%), 5 min each level; after removal, placed in xylene for 10 min, twice. The sections were mounted with neutral gum, and the slides were observed under an optical microscope.
Statistical analysis
All statistical data were analyzed using GraphPad Prism 9.0 software. Data are shown as the means ± SD of at least performed triplicate experiments. One-way Analysis of Variance (ANOVA) was used to determine the significance of statistical results for comparisons among multiple groups of data. The results were considered to be statistically significant when p values <0.05.
Results
Overview of sequencing data
Clean data from 10 samples were acquired after low-quality sequences were removed. Each sample had an average of 49.55 million reads and 14.86 billion bases (Table 3). When the Q30 ratio exceeded 94%, there was no observed GC bias, indicating that the high-quality sequencing data were of exceptional quality. Over 96% of the clean reads were accurately aligned to the rat reference genome (rn6), and 79.78-89.47% of the uniquely mapped reads were derived from the reads across all 10 samples. Principal component analysis revealed that the intragroup repeatability of the two groups of samples was high, and the differences between the groups were robust (Fig. 2A, C). The mRNA expression patterns of the samples demonstrated a strong correlation between the STC and NP (varied between 0.99 and 0.99), while the association between the two samples in one group was poor (Fig. 2B, D). These findings indicated that five samples within each group were successfully utilized for sequencing and that the results were consistent and representative of the downstream analysis.
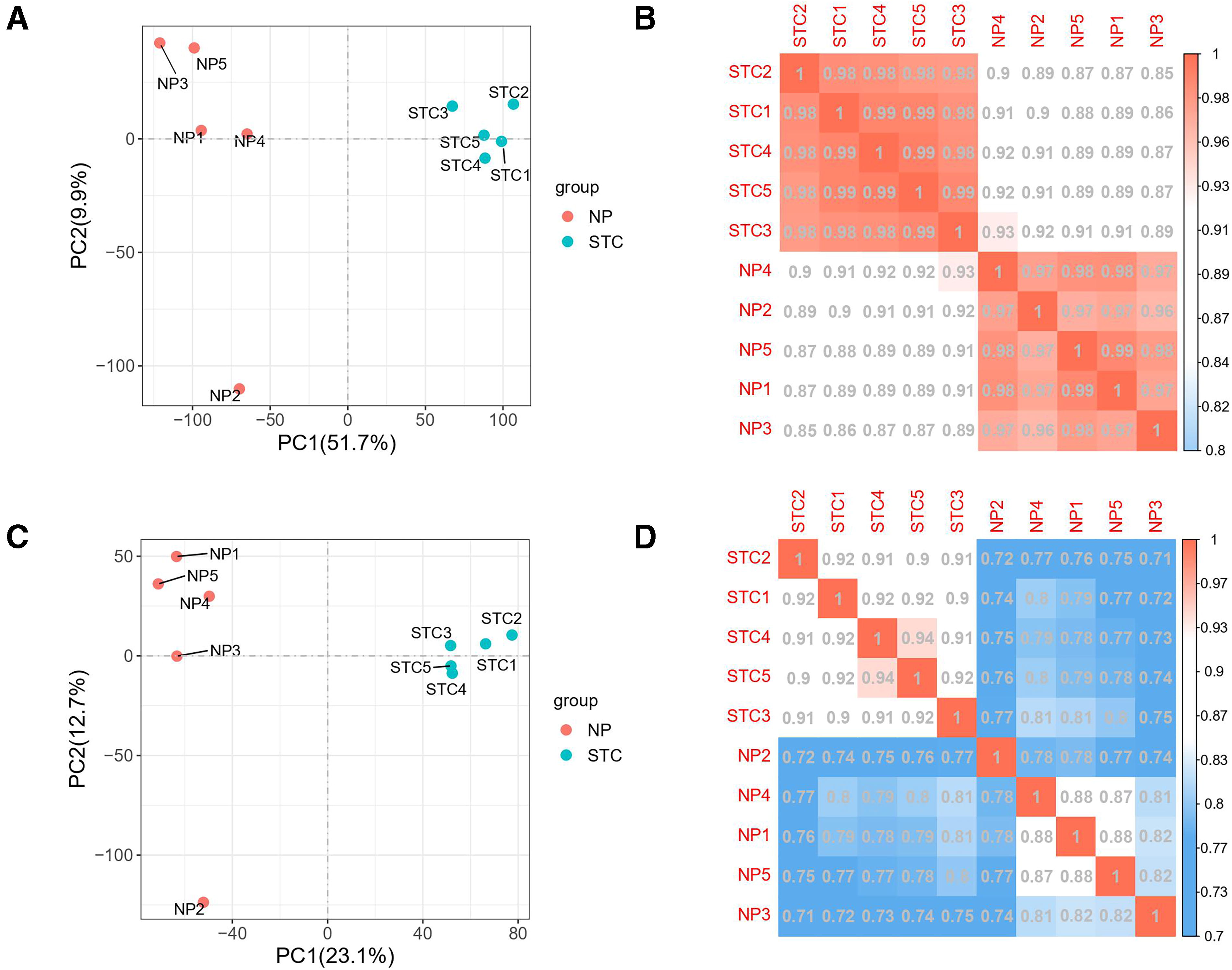
Relationship analysis between the STC and non-STC transcriptome samples.
Summary of RNA-Seq Data and Mapping
Identification of mRNAs differentially expressed in STC
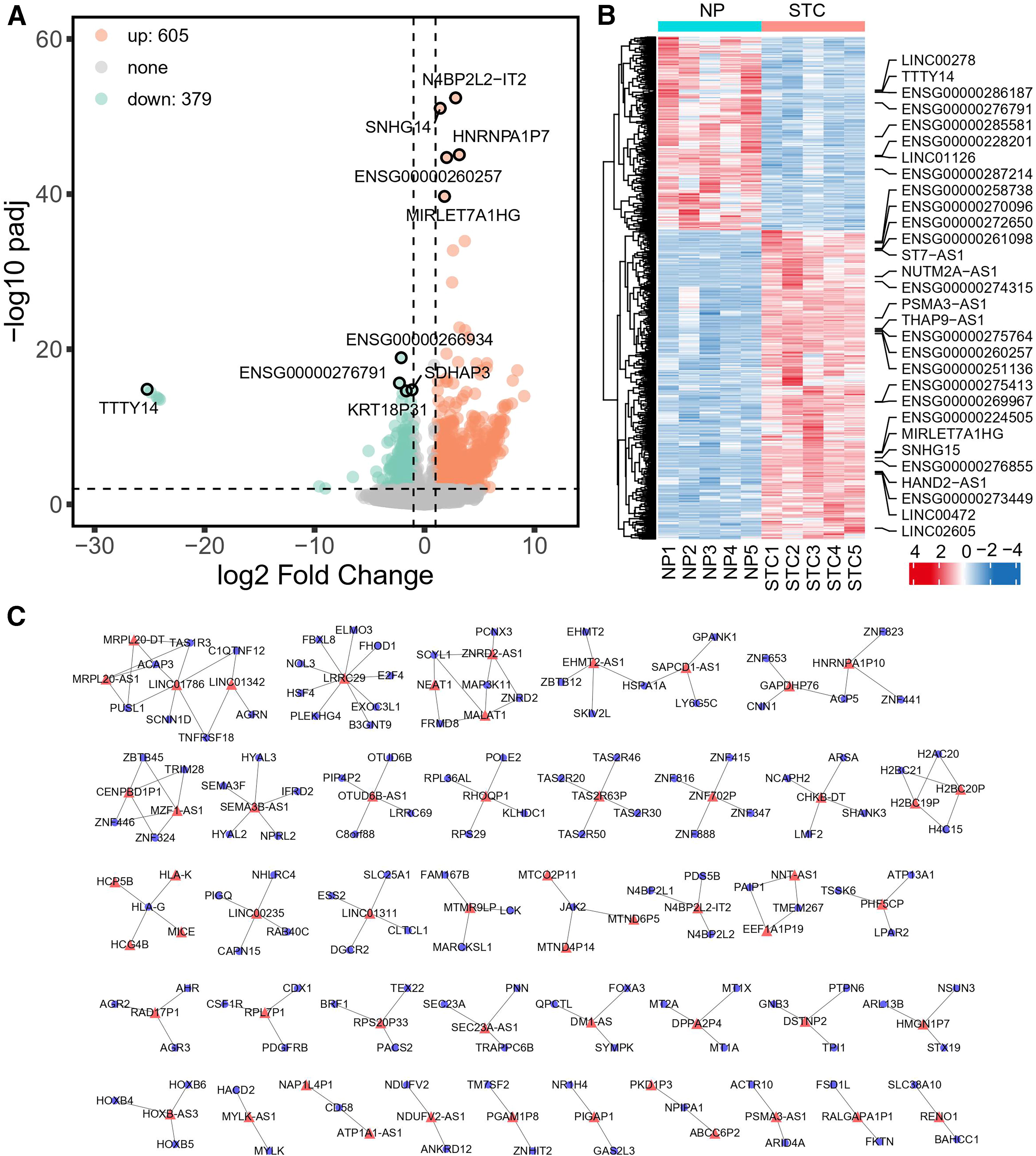
To analyze the differentially expressed genes (DEGs) in the two comparison groups, microarray technology was used. In total, 2880 mRNAs were upregulated, whereas 1550 mRNAs were downregulated (Fig. 3A). A heatmap displaying the top 30 DE mRNAs, ranked by fold change and encompassing both upregulated and downregulated genes, was constructed. (Fig. 3B). Protein network analysis was performed on the top 300 differential mRNAs to determine the potential regulatory links between mRNAs. The red mRNA was upregulated, whereas the green mRNA was downregulated. The top 300 genes were found to be dominated by upregulated genes, with hub genes such as POLR2B, SRSF1, and SUMO1 (Fig. 3C).

Differential expression analysis of mRNAs. Expression profiles and functional analysis of differentially expressed mRNAs.
GO and KEGG analysis of the DE mRNAs
The 15 GO terms of the DE RNAs are displayed in Figure 4. Concerning the GO analysis of the upregulated mRNA host genes, the most highly enriched GO terms were RNA localization, regulation of RNA splicing, DNA replication, and chromatin remodeling in biological processes (Fig. 4A).
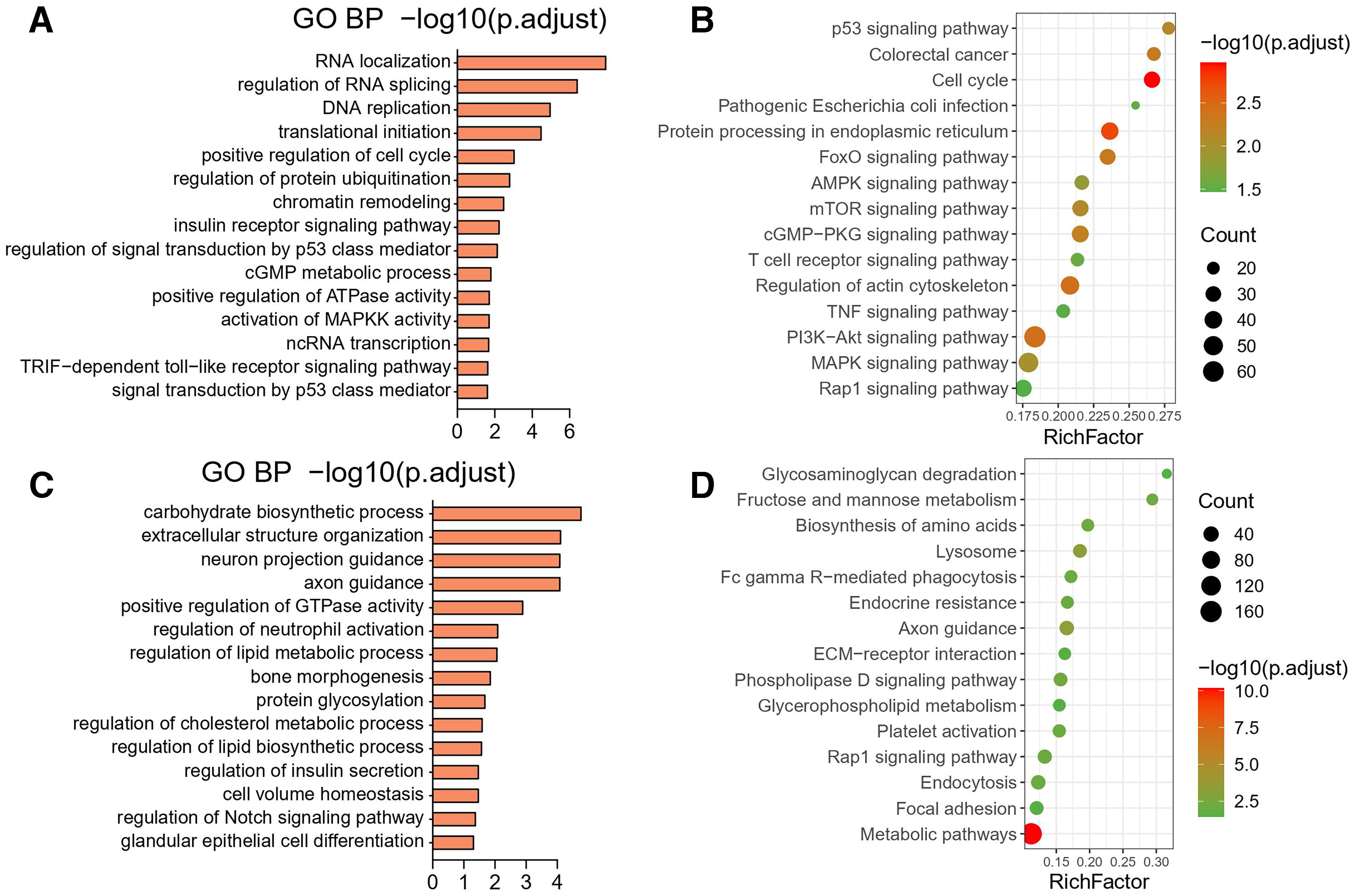
GO and KEGG analysis of dysregulated mRNAs.
Detection of lncRNA genes showing differential expression in STC
Analysis of the high-throughput RNA sequencing results from STC and control colon tissues resulted in the identification of lncRNAs. There were 984 DE lncRNAs, including 605 upregulated and 379 downregulated lncRNAs (Fig. 5A). Hierarchical heatmap clustering revealed pronounced distinctions in the lncRNA expression patterns between the two groups (Fig. 5B). We investigated the relationship between the altered lncRNAs and the differentially expressed mRNAs near 100 KB. We found that several lncRNAs may regulate multiple mRNAs. For example, LRRC29 may regulate nine adjacent mRNAs, including E2F4. SEMA3B-AS1 may regulate 5 mRNAs. These findings demonstrate that gene synthesis and protein translation are active processes. We also discovered that lncRNA transcription was substantial, indicating that lncRNAs could be associated with the STC process.
Analysis of the lncRNA expression profile in STC.
Coexpression of lncRNAs/mRNAs and function
The binding of miRNAs is a crucial function of lncRNAs in regulating mRNA expression. As a result, we developed a ceRNA mechanism. We selected the top 10 upregulated lncRNAs and the leading 1000 upregulated mRNAs and subsequently used miRanda (v3.3a, parameter-en-25) to predict lncRNA-bound miRNAs and TargetScan (v7.2) to predict miRNA-bound mRNAs. We found that JPX exhibited significant enrichment in the ceRNA network (Fig. 6A). Figure 6A provides information on the interaction pairs that were activated, including 5 upregulated lncRNAs, 15 upregulated miRNAs, and 379 upregulated mRNAs. Figure 6B shows the GO annotations indicating that numerous biological processes involving mRNAs were associated with the pidermal growth factor receptor (ERBB) signaling pathway and nuclear transport. According to the KEGG pathway analysis, the majority of the relevant pathways associated with the mRNAs were associated with the mitogen-activated protein kinase (MAPK) signaling pathway, autophagy-animal pathway, oxytocin signaling pathway, and cAMP signaling pathway (Fig. 6B). Figure 6C illustrates a repressed network that included 5 downregulated lncRNAs, 23 downregulated miRNAs, and 203 downregulated mRNAs. A significant portion of the BP host genes exhibited enrichment in the collagen metabolic process, activation of JUN kinase activity, regulation of epithelial-to-mesenchymal transition, and axon guidance.

The lncRNA-miRNAs-mRNAs networks and biological function and pathway analysis of differentially expressed mRNAs.
According to the KEGG signaling pathway analysis, host genes exhibited remarkable enrichment in five terms; axon guidance and the ECM-receptor interaction were the most significant (Fig. 6D).
Detection of altered expression of circRNAs within STC
The circRNA expression patterns in five colonic tissue samples from patients with STC and five non-STC samples were explored. In the STC group, a cumulative count of 14231 circRNAs was established, while in the non-STC group, a total of 6722 circRNAs were detected. The number of circRNAs on chromosomes 1, 2, and 3 exceeded that on all the other chromosomes, implying that these particular chromosomes may have a significant impact on the development of STC (Fig. 7A). Most genes can make 1-3 circRNAs. Only a handful of patients were capable of producing more (Fig. 7B). Analysis of sequence length indicated that the majority of circRNA splice lengths were 200-2000 nt (Fig. 7C). Within the pool of markedly altered circRNAs, 2059 were upregulated, and 93 were downregulated (Fig. 7D). Hierarchical heatmap clustering revealed clear disparities in the circRNA expression patterns between the two groups (Fig. 7E).
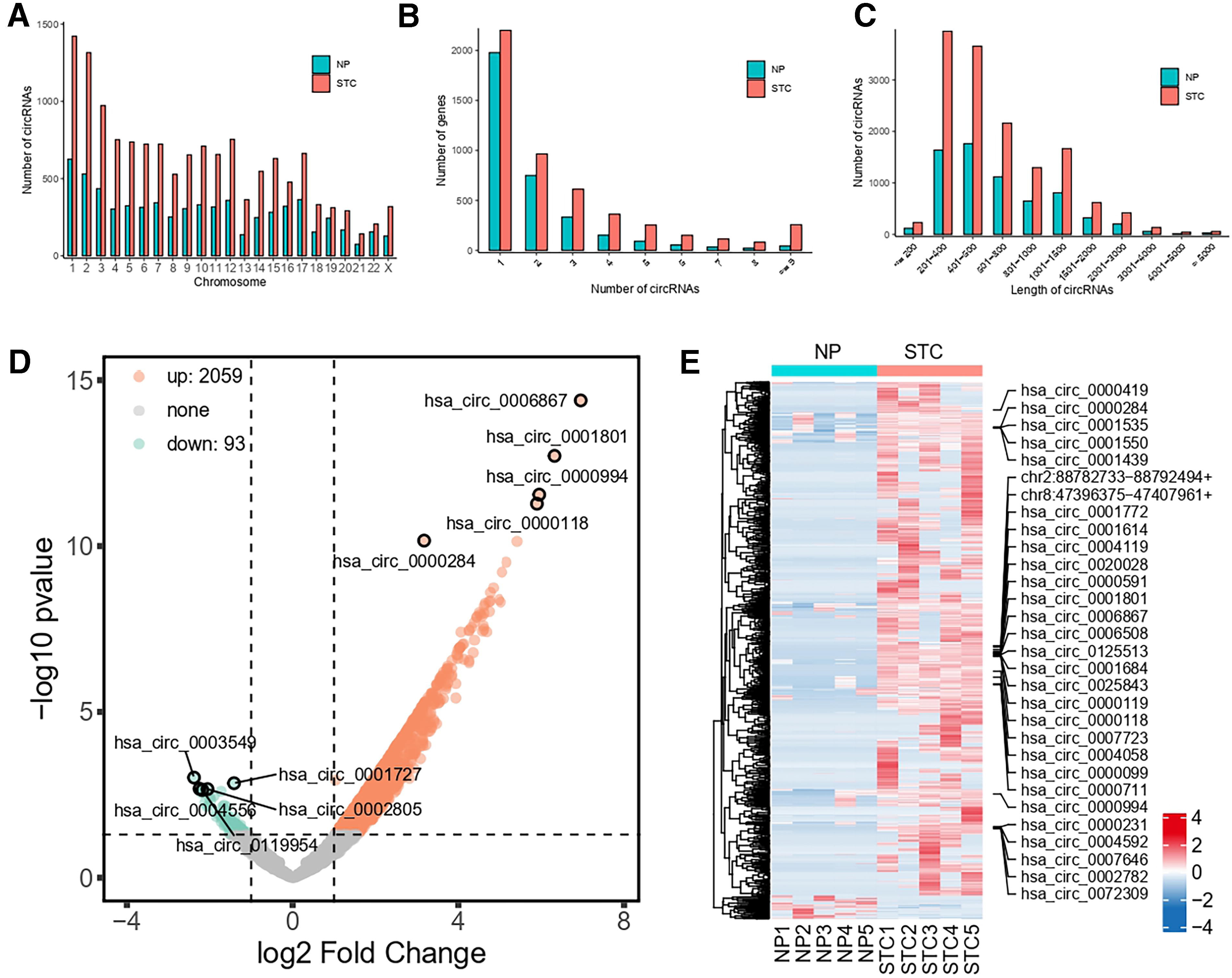
Gene expression characterization and expression profiles of differentially expressed circRNAs in STC transcriptome samples.
Construction of the ceRNA network
CircRNAs are ceRNAs that can potentially act as sponges for miRNAs, thereby alleviating the inhibitory effects of miRNAs on their target genes (Sailer, 2022). To determine the ceRNA roles of these circRNAs, we used miRanda to predict the interactions between variably expressed circRNAs and miRNAs. The results revealed 6 circRNAs, 17 miRNAs, and 395 target mRNAs, each of which represented a specific function. Additionally, the differentially expressed gene hsa_circ_0000994 was the key regulator of STC development. Hence, we constructed a regulatory network of circRNA-miRNA-mRNA interactions to elucidate the possible contributions of circRNAs to the modulation of STC between the two groups. Notably, within the ceRNA network, circRNAs were observed to contend for the same RNA entities to govern distinct sets of mRNAs. These RNA interactions offer a novel vantage for understanding the pathogenesis of STC (Fig. 8A). GO enrichment analysis indicated that the upregulated mRNAs associated with STC were primarily associated with biological processes such as the regulation of alternative mRNA splicing, the regulation of protein stability, and the regulation of RNA splicing (Fig. 8B). KEGG enrichment analysis revealed that the MAPK signaling pathway was the most notably enriched pathway (Fig. 8B). We selected the foremost 10 downregulated circRNAs and the uppermost 1000 downregulated mRNAs associated with STC to construct a coexpression network aimed at predicting the function of circRNAs (Fig. 8C). The results demonstrated that 5 circRNAs, 18 miRNAs, and 206 mRNAs were integrated into the coexpression network. One mRNA exhibited significant correlations with numerous circRNAs. Moreover, eight mRNAs were linked to other miRNAs through circRNAs, one of which was hsa_circ_0008699 (Fig. 8C). To clarify the possible role of circRNAs, GO function and KEGG pathway annotations were performed for these downregulated mRNAs. We identified the biological processes “extracellular matrix organization,” “activation of JUN kinase activity,” and “axon guidance.” The “protein digestion and absorption,” “ECM-receptor interaction,” and “TGF-beta signaling” pathways were found to be enriched in the KEGG pathway analysis (Fig. 8D).

The circRNA-miRNAs-mRNAs networks and biological function and pathway analysis of differentially expressed mRNAs.
STC model evaluation
The intestinal motility of mice in control and STC groups was evaluated by fecal particle count, fecal water content, and intestinal transit function, and it was found that compared with the control group, the fecal particle count, fecal water content, and intestinal transit function of mice in the STC model group was significantly reduced. Mice in the control group had complete mucosal, submucosal, muscularis, and outer membrane structures in their colonic tissue, according to histopathological analysis. The mucosa was covered in single columnar epithelial cells, and the lamina propria contained numerous intestinal glands. However, in the model group, the tubular structure was absent; only necrotic cell fragments and a small number of lymphocytes or neutrophils were visible, and certain areas of colonic tissue had mucosal necrosis, loss of epithelial cells in the necrotic area, and necrotic intestinal glands in the lamella propria. Semiquantitative assessment of general colonic structure, shedding of mucosal epithelial cells, and infiltration of inflammatory cells demonstrated that the pathological scores in the STC group were significantly higher, with a statistically significant difference when compared to the control group (p < 0.01). Constipation can result in heightened permeability of the colonic mucosa, a decline in goblet cell population, and diminished mucus production, ultimately weakening the intestinal mucus barrier. To visualize mucin-secreting goblet cells, Alcian blue staining was employed, which imparts a blue coloration to these cells. Compared with the control group, the number of goblet cells and mucus area in the colon tissue of rats in the STC group were markedly reduced (p < 0.01).
ICC are known to play a role in the development of various gastrointestinal motility disorders, such as idiopathic STC. c-Kit is recognized as a specific marker for ICC, facilitating their proliferation and growth through binding to the stem cell factor (SCF) receptor. Experimental findings indicate that c-Kit is predominantly localized in the apical and lateral regions of mucosal epithelial cells in colon tissue. In comparison to the control group, the STC group exhibited a decrease in the expression of c-Kit in the colon tissue of mice with constipation (Fig. 9). Taken together, these results indicate that Lop-induced STC mice.
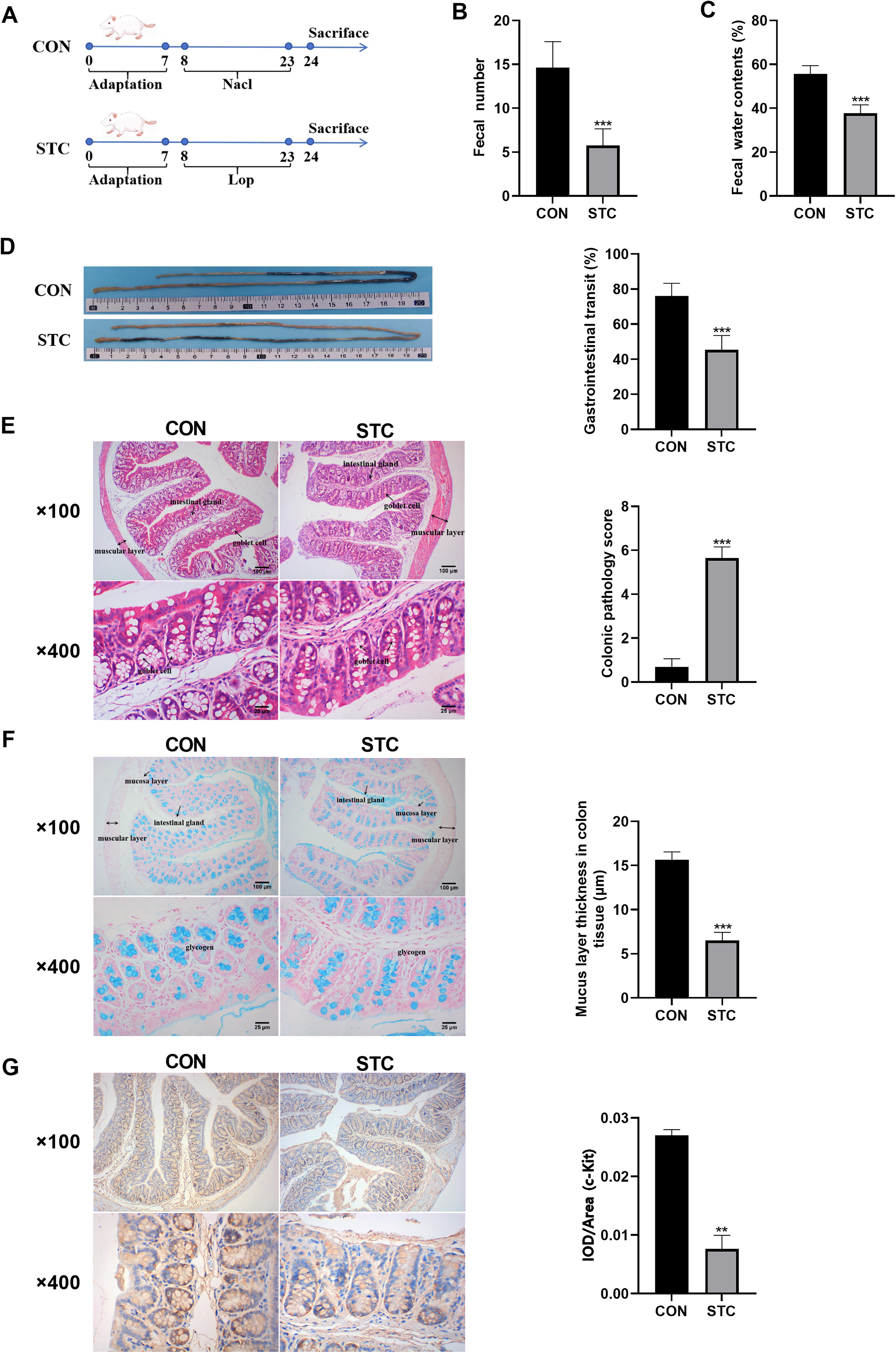
Construction and evaluation of slow transit constipation model in mice.
The expression of POLR2B, SRSF1, and SUMO1 in loperamide-induced constipation mice
We selected the top three DEGs for experimental validation on the established STC mice model. We found that SUMO1 expression in the colon of constipated mice was significantly lower than that of normal mice (p < 0.01) (Fig. 10). Conversely, the levels of SCF and c-Kit were no significant difference was observed between the control and the STC groups (p > 0.05) (Fig. 10).
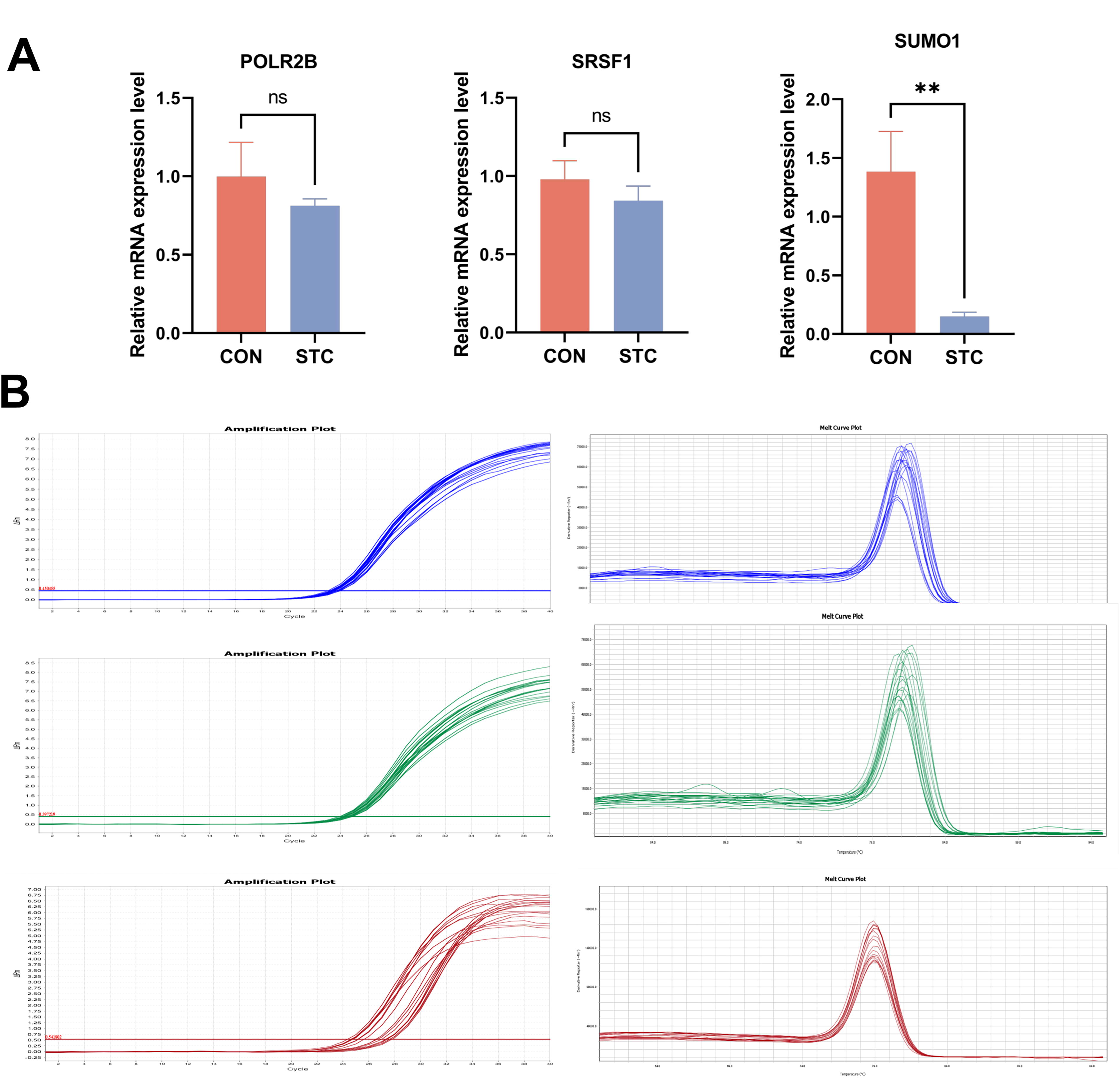
The mRNA expression levels of POLR2B, SRSF1, and SUMO1 in the colon in mice. Data represent the mean ± SD (n = 6). Statistical analysis was conducted using one-way ANOVA test for each group using GraphPad Prism 9.
The expression of POLR2B, SRSF1, and SUMO1 7in loperamide-induced constipation mice
We investigated the expression of POLR2B, SRSF1, and SUMO1 in the colons of mice with loperamide-induced constipation. As shown in Figure 11, the levels of SUMO1 were decreased in the STC group compared with the control group (p < 0.05), whereas the levels of POLR2B and SRSF1 were no significant statistical changes between the control group and the STC group.
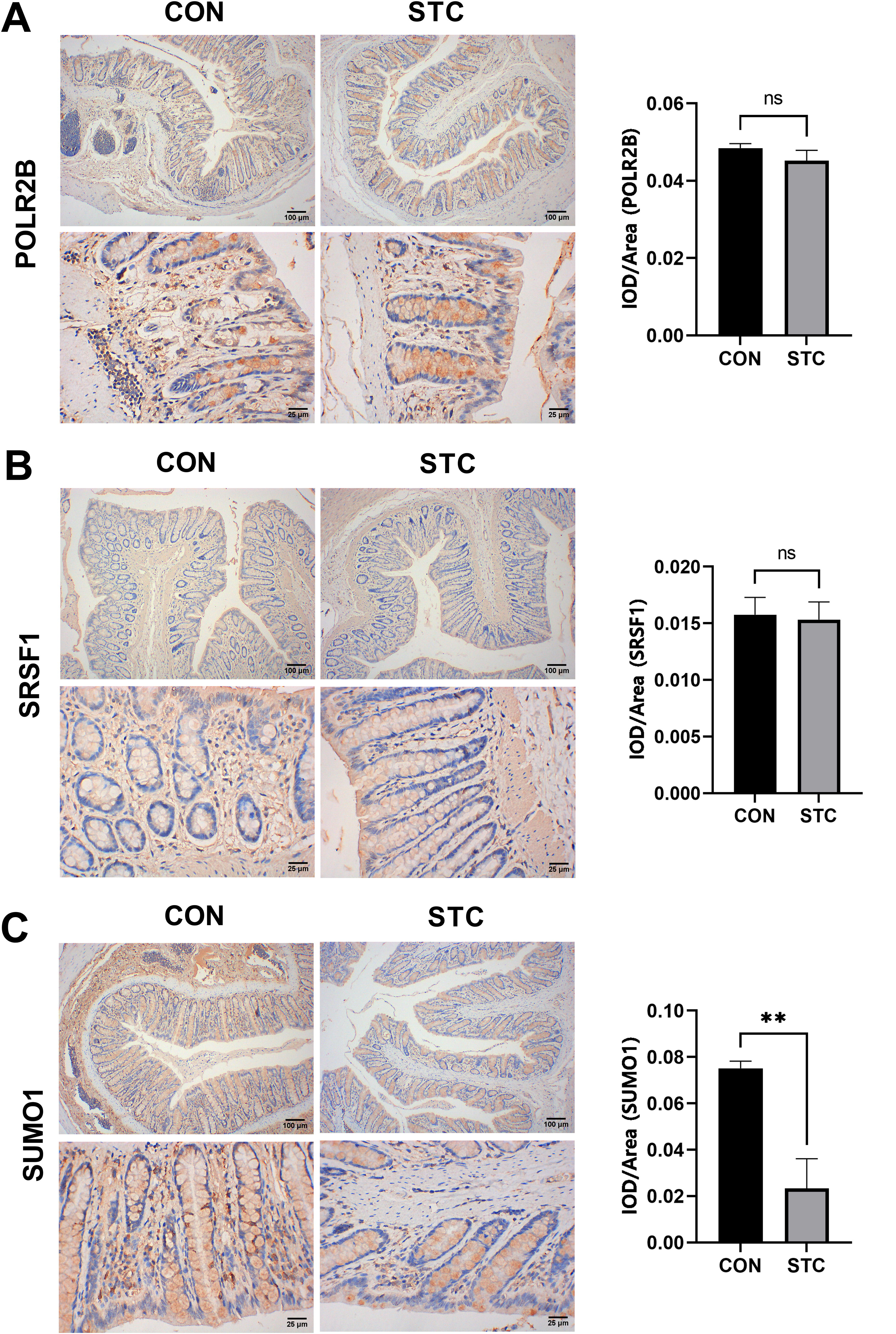
Protein expressions of POLR2B, SRSF1, and SUMO1 in the colons of mice were detected by immunohistochemistry analysis. The mean optical density (MOD) of POLR2B
Discussion
STC represents a fundamental functional disorder distinguished by reduced motility and poor colonic function and has an impact on patients’ health and quality of life (Sailer, 2022). For individuals with refractory STC unresponsive to medication, surgery could emerge as the sole viable choice (Chaichanavichkij et al., 2021). Many attempts have been made in recent years to investigate the molecular causes of STC. RNA-seq is a powerful method for elucidating the molecular functional characteristics of diverse organisms through transcriptome profiling (de Goede et al., 2021; Wang et al., 2022; Yang et al., 2021). There are currently few previous bioinformatics studies on STC, and STC genomic research is still in its early stages. Xi et al. (2022) discovered a significant correlation between hsa_circ_0004214 and muscle contraction modulation during the pathogenesis of STC. Furthermore, the results of Yu et al. (2020)demonstrated a correlation between a key miRNA (hsa-mir-619) and STC via weighted gene correlation network analysis. Other studies on STC have mostly involved animal experiments, with little investigation into chromosomal expression profiles (Geramizadeh et al., 2009; Kishi et al., 2020; Wang et al., 2024; Yan et al., 2021). Therefore, a better understanding of the pathophysiological mechanism behind STC will aid in its decline. However, the roles of circRNAs, lncRNAs, and mRNAs in colon specimens from STC patients are still unclear. Within this investigation, a set of transcripts with altered expression levels and certain essential variables were identified via comprehensive transcriptome sequencing analysis of colon specimens from STC patients, indicating that these transcripts could play an essential role at the microscopic level in STC.
Normal Cajal cell proliferation and intermuscular nerve system development are the foundations of animal gastrointestinal motility and can be modulated by numerous genes or the expression profiles of identical genes throughout various embryonic stages. Within this investigation, 4430 DE mRNAs, 984 DE lncRNAs, and 2152 DE circRNAs were detected via RNA sequencing in five constipated patient tissues as opposed to five colon cancer tissues compared with their paired normal tissues. After investigating the interactions between the observed genes with differential expression, we identified POLR2B, a hub gene that stimulates the process of transcription of DNA into mRNAs, snRNAs, and microRNA precursors (Yu et al., 2020). To explore the interaction between the detected DEGs, we constructed a PPI network, and we found the top three hub genes, POLR2B, SRSF1, and SUMO1, which play a pivotal role in the pathological process of STC. To verify the reliability of the results of three key genes, their expression levels in the colon tissue of STC mouse model were validated by RT qPCR. The results showed a significant decrease in the expression of SUMO1 STC in colon tissue. In order to further verify the protein expression levels of three key genes in colon tissue, immunohistochemical detection was performed. The expression of SUMO1 protein was significantly decreased in the colon tissue of STC mice.
In post-translational modifications of proteins, small ubiquitin-related modifier proteins (SUMOs) belong to a crucial class of regulatory proteins. Since its first proposal, SUMO modification has played an indispensable role in biology missing roles. As pacing cells, regulators, and receptors in the gastrointestinal tract, interstitial cells of Cajal (ICC) mediate the transmission of electrical activity, participate in signal transduction between the enteric nervous system and smooth muscle cells in the colon, regulate neurotransmitters, and play an important role in the generation, maintenance, and regulation of gastrointestinal motility. Geramizadeh et al. (2009) found through HE staining after colon resection in STC patients that the number of ICC in the inner and outer longitudinal muscle layers, as well as the intermuscular nerve plexus of the colon, was significantly reduced compared to normal colon tissue. The volume of ICC was significantly reduced, the number of cell processes decreased, ganglion vacuolar degeneration, nuclear condensation, and ICC cell protrusions and connections were disordered. Torihashi et al. injected c-Kit receptor blocker ACK2 into newborn mice for 8 consecutive days, causing the disappearance of ICC cells in the small intestine. The morphology and structure of cells in the residual c-Kit positive pacing area were similar to smooth muscle cells, leading to colonic motility disorders (Kishi et al., 2020). To meet the energy requirements of different organs in the body, mitochondria maintain dynamic stability in morphology, structure, quantity, volume, mass, and function through a series of mechanisms, known as mitochondrial homeostasis, which is the basis for ensuring the normal operation of energy metabolism. A study based on a rat model of gastrointestinal motility disorders reported that the ICC in the lower esophagus, gastric motility pacing area, small intestine, and colon tissues exhibited cellular damage changes such as mitochondrial degeneration and cytoplasmic dissolution, resulting in an increase in gastric residual rate and a decrease in small intestine propulsion rate (Wang et al., 2024). Zheng et al. observed swelling, vacuolization, myelin-like changes, and even dissolution of ICC mitochondria in the colon of STC rats (Zheng et al., 2021). They also observed a decrease in cristae, increased division, and an increase in the number of lysosomes, with a small amount of autophagosomes visible. Therefore, it is speculated that the imbalance of mitochondrial homeostasis in ICC may be a key factor in the abnormal quantity, structure, and function of ICC, which in turn triggers colonic motility disorders. In summary, restoring mitochondrial homeostasis is highly likely to be a prerequisite for maintaining normal development and proliferation of ICC and an important strategy for treating STC. Therefore, in-depth research on the relationship between regulating mitochondrial homeostasis and the abnormal phenotype of ICC cells has become a potential entry point for preventing and treating STC. In the rat STC model and gastrointestinal motility disorder model, PINK1 and Parkin protein and mRNA levels were increased in gastrointestinal tissue expression, ICC mitochondrial autophagy levels were elevated, and gastrointestinal transport dysfunction was aggravated (Wang et al., 2024; Zhang et al., 2022). An in vitro cell experiment demonstrated that MUL1-induced SUMO2 accumulation promoted SUMOylation modification of the K262 amino acid site of NDP52, inducing PINK1/Parkin-mediated mitochondrial autophagy (Gao et al., 2024). SUMO1 is involved in the regulation of numerous cellular processes, such as overseeing DNA damage repair and managing mitochondrial dynamics (Mazzone et al., 2020), and is a key element in the progression of STC. It is conventionally believed that SUMO1, which serves as a crucial mediator of presynaptic neurotransmitter release, modulates the performance of the CaV2.2 (N-type) voltage-gated calcium channel (Xu et al., 2021). In addition, when osmotic stress or methyltransferase activity is inhibited, SUMO-1 is localized within neuronal Cajal bodies in adult nervous tissue (Zheng et al., 2021). The reversible SUMOylation process acts as a critical link connecting mitochondrial dynamics (fission/fusion) and mitophagy, collectively maintaining mitochondrial homeostasis. This molecular mechanism provides a scientific foundation for investigating the recovery pathways of colonic motility disorders in STC and developing potential therapeutic interventions. Although experimental validation has shown that SUMO1 is significantly downregulated in the colon tissue of STC mice, larger clinical studies are still needed to confirm these findings.
While our RT-PCR and Western blot analyses in the loperamide-induced STC mouse model strengthened the reliability of the bioinformatics findings, we acknowledge that molecular mechanisms underlying STC may differ between mice and humans. For instance, species-specific variations in gene regulatory networks or immune-microenvironment interactions could influence translational applicability. Future studies should prioritize validating these results in human STC tissues, particularly given the heterogeneity of clinical samples and potential confounding factors in human patients. We explicitly recommend that future work prioritize validation in human STC tissues, particularly for hub genes (POLR2B, SUMO1, SRSF1) and the ceRNA network, to confirm their clinical relevance. We note that ethical and practical challenges (e.g., scarcity of fresh human STC tissues, heterogeneity in patient samples) currently limit such validation but underscore its importance for therapeutic development.
Conclusion
This is the first study in which high-throughput sequencing of tissues from STC patients and non-STC patients was performed to determine the true genetic basis of constipation. Moreover, SUMO1 were found to be potential critical gene markers that play a role in the development of STC in patients. This study has several limitations. First, RNA sequencing was performed on a small number of samples; case selection bias may be introduced and affect the accuracy of the results. Second, although expression validation was conducted at the animal level, the sample size was relatively limited. Third, although SUMO1 has been validated through Quantitative Real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry, more studies, including Western blot, are needed to further confirm these results. Therefore, further validation with more clinical cases is needed to confirm the validity of the findings.
In summary, this study not only deepens our understanding of STC, a common digestive system disease but also provides new possibilities for personalized treatment and early diagnosis. Future research needs to further validate the potential applications of these biomarkers in a larger population of patients and explore the yet to be elucidated molecular mechanisms and potential therapeutic targets.
Footnotes
Authors’ Contributions
M.S. and Y.W. wrote the initial draft of the article, enrolled patients in the study, and assisted in the interpretation of the data. H.Y. and X.W. contributed to the development of the article. L.S. conducted the qRT-PCR and immunohistochemistry experiments. S.Y. designed the study. All the authors approved the final version of the article.
Data Availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request. All the raw sequencing data were deposited in the NCBI Short Read Archive database under the GEO repository ID GSE245885.
Author Disclosure Statement
All the authors declare that they have no competing interests.
Funding Information
This work was supported by the Medical Research Project of Jiangsu Provincial Health Commission (Grant No. M2022104), the Suzhou Medical and Health Science and Technology Innovation Project (Grant No. SYW2024032).