
Abstract
Abstract
Introduction
Case
A 14-month-old girl was admitted to the hospital with a history of abdominal pain and vomiting. There was no history of passing blood in stool or urine. Physical examination revealed a palpable mass measuring 12×10 cm in the right pelvic region. Findings from other systemic examinations were normal. Laboratory investigations were as follows: hemoglobin 10 g%, microcytic hypochromic picture and white blood cell count 6000/mm3. Urine analysis was normal and all biochemical investigations were within normal limits. Tumor markers (alpha-fetoprotein and beta human chorionic gonadotropic [β-hCG]) were within normal range. On ultrasonography, bilateral kidneys, liver, gallbladder, spleen, and pancreas were normal. Contrast-enhanced computed tomorgraphy (CECT) scan was performed using non-ionic contrast medium, and it showed a heterogeneously enhancing lesion in the right pelvis and lower abdomen measuring 12×12cm. Both the kidneys were of normal size and density, and excreting concentrating contrast normally. Perirenal fascial planes were normal. Bilateral adrenal glands, liver, gallbladder, pancreas, and spleen were normal. No free fluid was seen, and a final diagnosis of heterogeneously enhancing lesion in the pelvis and lower abdomen of ovarian origin was given on CECT. The patient was taken for exploratory laprarotomy. Perioperatively, a 12×12 cm lobulated intra-abdominal mass was identified arising from the ovary. It was not adherent to surrounding organs. Complete surgical excision of the mass was performed, in addition to removal of right ovary and tube.
Pathologic findings
Grossly, the excised tumor was binodular, measuring 10×8 cm with solid and cystic areas, and was associated with foci of hemorrhage and necrosis (Fig. 1). The ovary could not be seen separately. The fallopian tube was not involved. Microscopically, the tumor showed a mixture of undifferentiated blastema, and epithelial and mesenchymal tissue. The blastematous areas were extremely cellular and composed of small round-to-oval cells admixed with primitive epithelial tubules and scattered glomeruloid structures. Blastema and epithelial elements were separated by stroma consisting of spindle cells of indeterminate type. There was no evidence of anaplasia. Teratomatous elements were not seen, despite exhaustive sampling. In only one of the sections was a peripheral rim of residual ovarian tissue seen (Fig. 2). Immunohistochemistry was performed. Tubules were positive for keratin and stromal cells were positive for vimentin (Fig. 3). A diagnosis of primary ovarian Wilms' tumor was made.
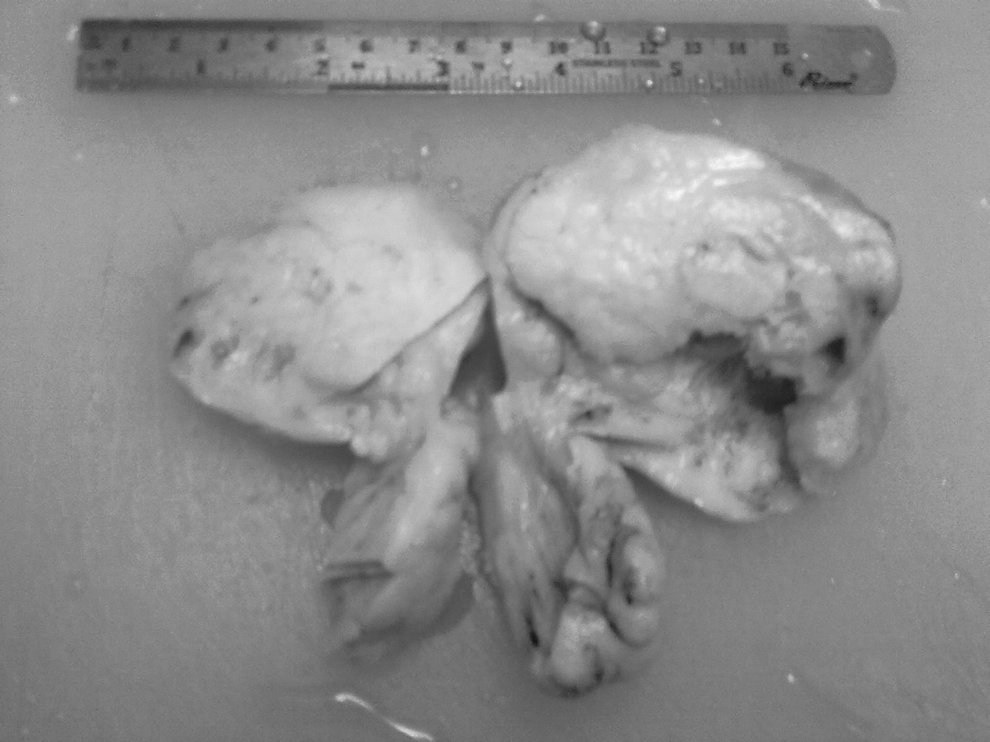
Binodular tumor; cut section is gray-white with areas of hemorrhage and cystic degeneration.

Wilms' tumor showing characteristic triphasic histology, including epithelial (tubules and glomeruli), blastemal, and mesenchymal components (

Immunohistochemical (IHC) staining: Epithelial memberane antigen (EMA) positivity in epithelial component (
Postoperative period
The postoperative period was uneventful and patient was referred to pediatric oncology for further treatment. She received four cycles of chemotherapy, which included two drugs, vincristine and actinomycin, and has been on regular follow up since then.
Discussion
Wilms' tumor is a malignant tumor with a characteristic histologic appearance, seen typically in children and almost exclusively involving the kidney. ERWT are exceedingly rare and may be seen in the inguinal region, endocervix, uterus, epididymis, ovary, testis, and any place in retroperitoneum along paravertebral area. The age at presentation usually ranges from 2 months to 10 years. 3 Their histogenesis, morphology, clinical staging, behavior, prognosis, and response to therapy are similar to those of renal Wilms' tumor.
The origin of ERWT is controversial and unclear, as this condition is very rare. However there are different schools of thoughts including
1. Origin from metanephric blastema, as the majority of the tumors occur in the retroperitoneal region. However, the presence of ERWT cephalad to the kidney refutes this hypothesis. 4
2. Origin from primitive mesodermal tissue; occurrence of ERWT in the cervix, vagina, and inguinal canal support this theory. 5
3. Connheim's cell rest theory: according to this hypothesis, origin of tumor is from a cell with persistent embryonal potential to undergo malignant transformation at any point in time. 6
Based on their probable histogenesis, ERWT can occur in two settings. First, those arising within teratomas; and second, those in which there is no evidence of a teratomatous origin. 1 Histogenesis of the second type is controversial.
While diagnosing a tumor as ERWT, extension from the intrarenal tumor should be ruled out and other differential diagnosis should also be considered. In this case, there was no evidence of primary renal tumor either by imaging or perioperatively. The most important differential diagnosis is immature teratoma composed largely of nephroblatic tissue. No teratomatous element was identified in this case, despite exhaustive sampling. The second possibility could be retiform Sertoli–Leydig cell tumor because of the presence of tubular structures. However, the presence of blastemal tissue and the absence of typical Sertoli–Leydig areas excluded this possibility. Adenocarcinoma and malignant mixed mullerian tumor should also be included as a differential diagnosis and they can be distinguished from Wilm's tumor by the presence of typical carcinomatous foci and the absence of blastemal and glomeruloid elements.
Staging and management of extrarenal nephroblastoma is the same as for its renal counterpart. 7 All cases are treated by surgery with postoperative adjuvant chemotherapy. Radiotherapy is reserved for unresectable tumors and those with distant metastasis. The presence of anaplasia indicates poor prognosis with increased resistance to chemotherapy. 8 In this case, features of anaplasia (polyploidy and nuclear and mitotic atypia) were not seen. The patient was treated surgically with postoperative adjuvant chemotherapy and is on regular follow-up.
Conclusions
Although very rare, Wilms' tumor may occur in sites other than the kidney and should also be considered as one of the differential diagnoses of an abdominal mass in children. Morphology, behavior, prognosis, and response to therapy of this tumor are similar to those of renal Wilms' tumor; therefore, similar staging and treatment protocol can be followed.
Footnotes
Disclosure Statement
No competing financial interests exist.