
Abstract
Abstract
Background:
Caudal duplication syndrome is a rare entity characterized by complete duplication of the genitourinary system and hindgut structures. There have been 13 case reports of women with genitourinary duplication; only 3 had complete caudal duplication and only 1 had a successful term pregnancy. There are no case reports to date of patients having more than 1 successful pregnancy with this syndrome.
Case:
A 28-year-old multiparous woman with known caudal duplication syndrome presented with recurrent urinary tract infections and hydronephrosis. This patient was interested in having a unification of her two vaginas. An examination under anesthesia revealed two gluteal clefts, three labia minora, two vaginas widely separated by >4 cm, a bifid clitoris, two normal cervices, two urethras, a right perforate anus, and a left imperforate anus. Exploratory laparotomy revealed uterine didelphys, 2 bladders, 2 ureters, and 2 kidneys.
Results:
Excision of the left bladder and ureteral reimplantation resulted in complete resolution of her hydronephrosis. Vaginoplasty was not recommended in this case, because the separation between the vaginas was >4 cm.
Conclusions:
Caudal duplication syndrome is an extremely rare condition most often recognized in utero, at birth, or during infancy, with complete duplication of the genitourinary and hindgut structures. Many of these patients have the ability to achieve successful gestation, but have an elevated risk of pregnancy and/or urologic complications. Collaboration with a gastroenterologist, urologist, and gynecologist is critical for optimizing care. (J GYNECOL SURG 33:153)
Introduction
C
Caudal duplication syndrome, a term coined in 1993, is an extremely rare entity characterized by complete duplication of the genitourinary system and hindgut structures and is thought to arise at ∼25 days of gestation. 4 Most cases are diagnosed in infancy, although some are not recognized until adulthood. There have been 13 case reports of women with genitourinary duplication, only 3 having complete caudal duplication and only 1 having a successful term pregnancy. 5 In addition to obstetric risks, urologic and gastrointestinal complications have been described, including urinary and fecal incontinence, vesicoureteral reflux, recurrent urinary tract infections (UTIs), hydronephrosis, and recurrent rectovaginal fistulas. 6 It is imperative to identify these malformations accurately in order to detect associated anomalies, avoid unnecessary surgeries, and counsel the patients better on their fertility potentials and risks.
Case
A 28-year-old Hispanic female presented to the University of Texas Southwestern Medical Center, in Dallas, TX, with recurrent UTIs—this was her third presentation in the last year for a culture-proven UTI. She also reported similar prior presentations for UTIs and pyelonephritis. This patient had a history of caudal duplication syndrome that was diagnosed at birth. She also had undergone an exploratory laparotomy in Mexico at the age of 4. She was found to have a duplicated colon with one imperforate and one stenotic anus, and subsequently underwent anastomosis and dilation of her stenotic anus. Later in life, she had two term cesarean sections from her right uterine horn as well as a bilateral tubal ligation. This is the first case to have more than 1 successful pregnancy with this syndrome. Upon presentation to the Medical Center, a computed tomography scan and an MRI of her pelvis showed a duplicated and trabeculated left urinary bladder with bladder-wall thickening, hydroureter, and mild hydronephrosis. A Lasix renogram revealed 27% and 73% function of the left and right kidneys, respectively.
Results
After urodynamic testing revealed poor compliance, detrusor overactivity, and incomplete emptying secondary to detrusor hypocontractility, the Medical Center's Urology team recommended starting clean intermittent catheterization (CIC) prior to surgery. As she was able to successfully perform CIC with her native right urethra, she did not need a catheterizable channel. She was taken to the operating room by the Medical Center's Reproductive Endocrinology and Infertility, and the Urology teams for an examination under anesthesia (EUA) and an exploratory laparotomy. The EUA revealed that she had two gluteal clefts, three labia minora, two vaginas widely separated by >4 cm (Fig. 1), a bifid clitoris (Fig. 2), two normal cervices, two urethras, a right perforate anus, and a left imperforate anus (Fig. 3). The exploratory laparotomy revealed uterine didelphys (Fig. 4) with the right horn slightly larger than the left one, bilateral fallopian tubes with surrounding scarring, two normal ovaries, two bladders, and a duplicated colon. Urology performed a left cystectomy with reimplantation of the left ureter into the right bladder, augmentation ileocystoplasty, and ureteral stent and suprapubic catheter placement for temporary urinary diversion. The suprapubic catheter was removed due to discomfort, and she remained continent with self-catheterizations and without any further UTIs. The hydronephrosis was completely resolved as noted on a renal US 1 month after the procedure. Although this patient had expressed interest in unification of her two vaginal cavities, vaginoplasty was not recommended in this case because the separation between the vaginas was >4 cm.

Two gluteal clefts, three labia minora, two vaginas.
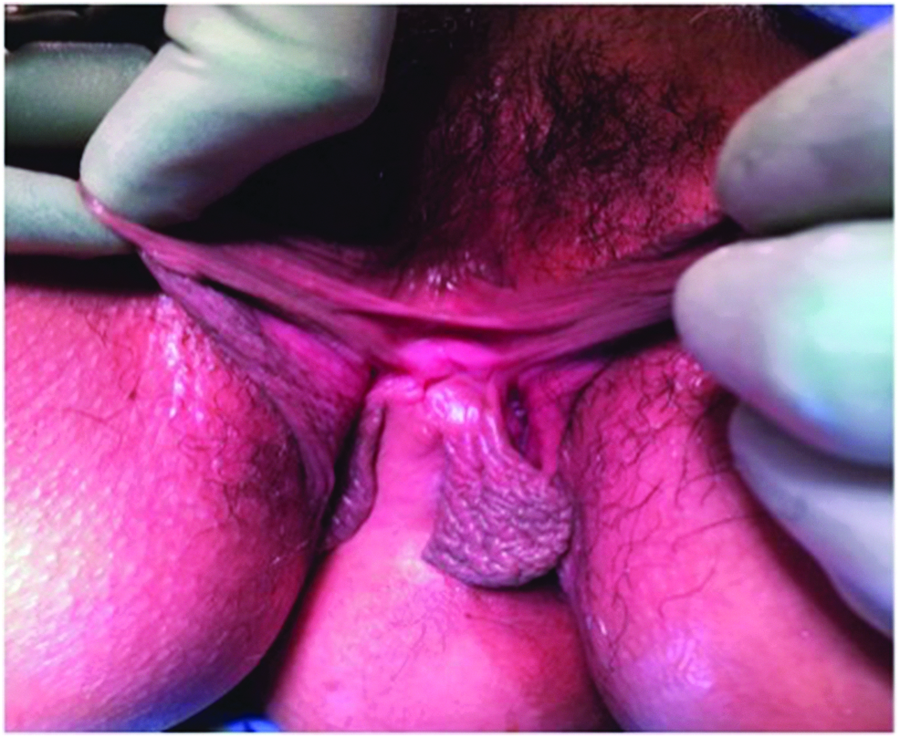
Bifid clitoris, three labia minora.
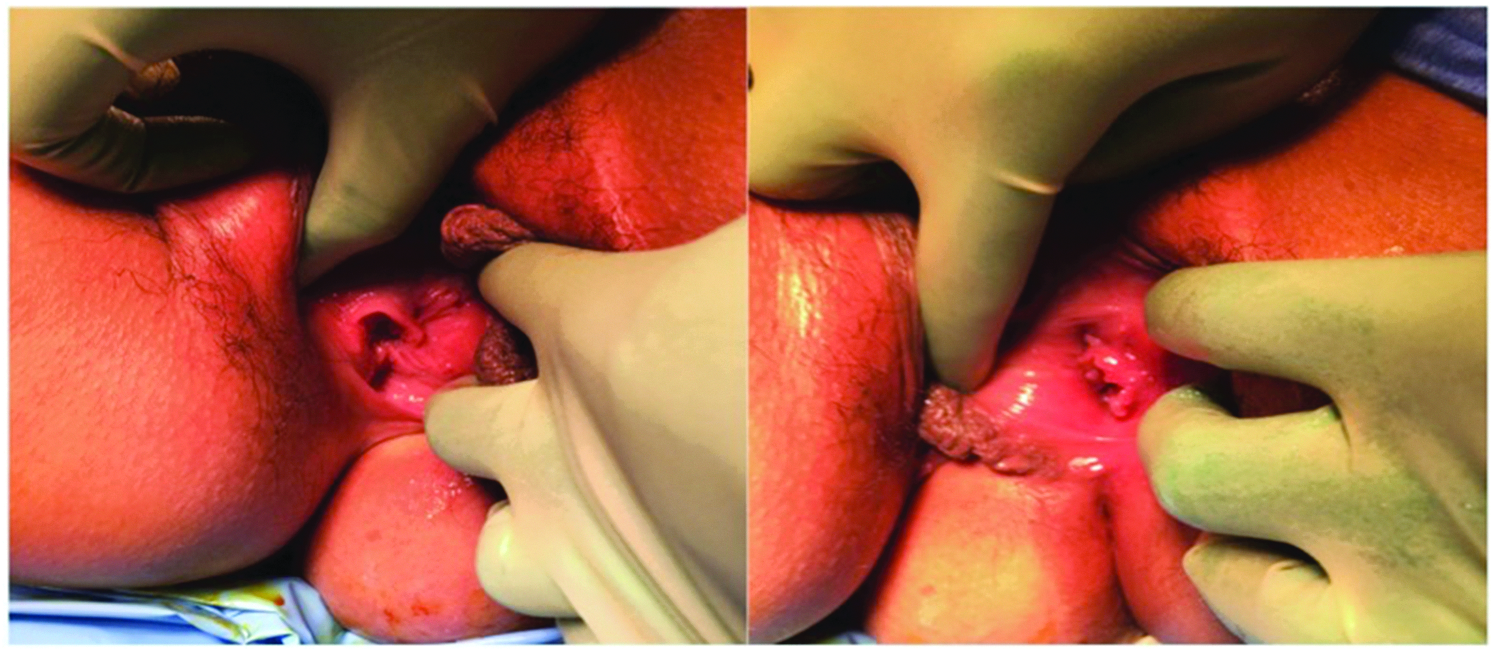
Right vagina and anus (left image); left vagina with no anus (right image).
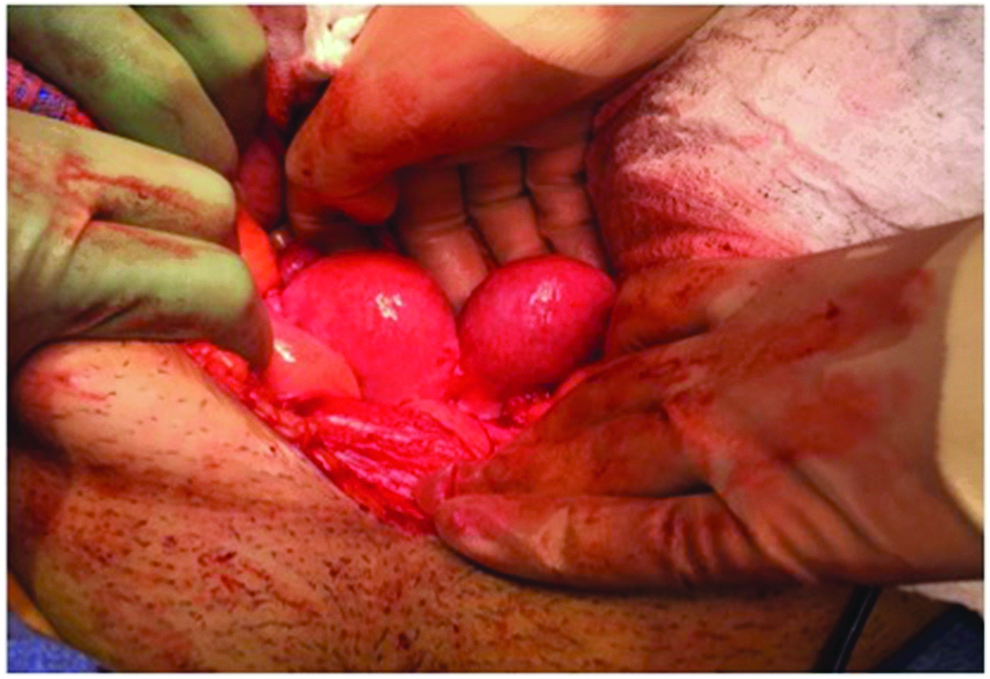
Uterine didelphys.
Discussion
A complete understanding of embryogenesis is paramount in the treatment of patients with congenital anomalies. In this patient, the midgut structures distal to the vitelline duct (including the distal ileum, cecum, appendix, and the proximal 2/3 of the colon), the hindgut structures (from the distal 1/3 of the transverse colon to the anal canal), and distal hindgut structures of the cloaca that develop into the urogenital sinus (bladder, urethra, and vagina) were duplicated. She also had incomplete fusion of the Müllerian ducts, resulting in uterine didelphys. The etiology of this syndrome is not well-understood. Theories include abnormal expression of distal HOX genes, AXIN1 genes in the WNT pathway, incomplete monozygotic twinning, and abnormal vacuolization of the hindgut.7–9
Conclusions
Other case reports have described surgical interventions, most often in infants usually involving reanastamosis of the duplicated colons. Few case reports have described surgical intervention for treatment of urinary complications, including incontinence and recurrent UTIs. Surgery typically describes resection of a bladder septum with resolution of symptoms.6,10 This current case report described excision of one bladder resulting in complete symptom relief.
The role of the obstetrician–gynecologist is the early delineation of the reproductive anatomy to counsel a patient and her family better about possible reconstructive options and future fertility. A uterine didelphys will put a patient at risk for recurrent miscarriages and preterm labor. Recurrent UTIs in these women should elicit prompt urologic evaluation and treatment to avoid permanent kidney damage.
Footnotes
Author Disclosure Statement
No competing financial conflicts exist.