
Abstract
Abstract
Background:
Aromatase and P450 oxidoreductase deficiencies are disorders of steroidogenesis, with a phenotypic spectrum ranging from ambiguous genitalia to Antley–Bixler syndrome. The aromatase complex affects estrogen synthesis and androgen metabolism and maintains the balance of the androgen–estrogen ratio in different tissues. Due to a lack of aromatase, high levels of ovarian androgens and gonadotropins facilitate ovarian cyst formation.
Case:
A 16-year-old patient was admitted to a clinic with acute lower abdominal and pelvic pain. Her menarche had occurred at 12 years of age and continued with irregular menses. She had partial labial fusion and clitoromegaly (Prader classification 2), with Tanner stage 3 pubertal development. A pelvic ultrasound showed enlarged and multicystic ovaries on both sides, with torsion of the right ovary. Laboratory investigations of marker levels revealed high follicle-stimulating hormone (31.28 mIU/mL; normal range: 1.7–7.7 mIU/mL), high luteinizing hormone (32.35 mIU/mL; normal range: 1–11.4 mIU/mL), and low estradiol (<5 pg/mL; normal range: 22.3–341 pg/mL).
Results:
Detorsion of the right ovary was performed via laparoscopy. During puberty and adulthood, daily estrogen and progesterone therapy was needed to facilitate breast development, maintenance of menses, bone mineralization, fusing of the epiphysis, and decreasing levels of gonadotropins.
Conclusions:
The current patient was diagnosed as having an aromatase deficiency with these findings but this could not be proven via genetic evaluation because of this patient's nonattendance. The decision of performing a cystectomy in a younger age woman should be considered very carefully. (J GYNECOL SURG 33:164)
Introductıon
H
After puberty, abnormal sexual development and virilization due to excess amounts of androgens can be seen. Aromatase deficiency is an autosomal recessive disorder due to mutations of the CYP19A1 gene. There have been ∼33 cases (23 females, 10 males)1–17 of aromatase deficiency reported. Six cases are summarized in Table 1,1–6 including the original case reported by Harada and colleagues in 1992. 1
BMD, bone mineral density; FSH, follicle-stimulating hormone; LH, luteinizing hormone.
Case
A 16-year-old patient was admitted to a clinic with acute lower abdominal pain, nausea, and vomiting that had lasted for 2 days. The severity of her pain had advanced during the last 6 hours prior to her admission. Her menarche had occurred at age 12, and she had irregular menses with an ∼40-day cycle, with 6 days of menstrual flow. Her last menstruation had occurred 3 weeks prior to her admission. She had clitoromegaly (Fig. 1; Prader classification 2) and partial labial fusion (Fig. 2), with Tanner stage-3 breast and pubic hair development. Her Ferriman Gallwey Score was 6. Her clitoromegaly was evident at birth but her family had not agreed to any investigations at that time.

Clitoromegaly.
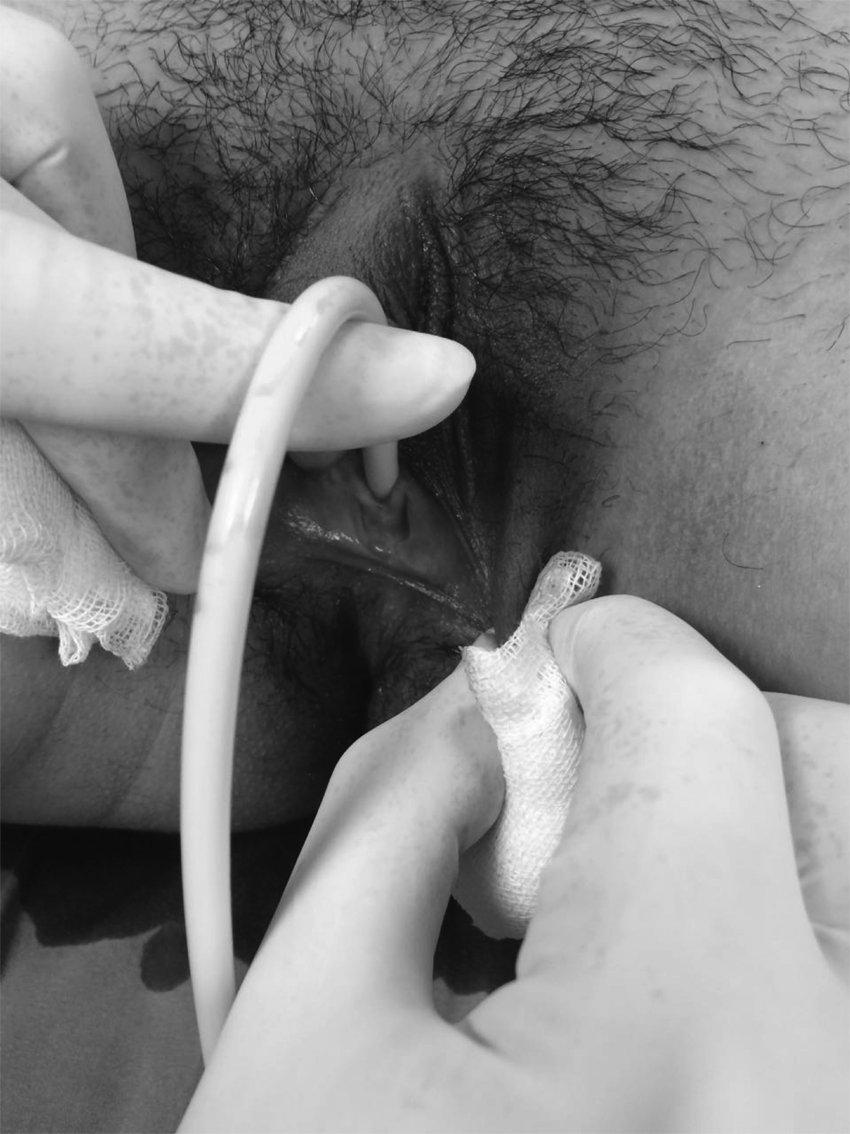
Partial labial fusion.
A pelvic ultrasound (US) showed enlarged and multicystic ovaries on both sides. The ovary on the right side was larger in diameter (82 × 49 × 36 mm), and her uterus appeared to be hypoplastic (60 × 20 × 10 mm). Doppler findings on the right ovary favored the possibility of torsion. This patient was not receiving ovarian stimulation treatments. She was diagnosed as having right ovarian torsion and was taken to the operating room for further treatment. A laparoscopy was performed and showed an enlarged and edematous 720° rotated right ovary (Fig. 3). Her uterus was hypoplastic (Fig. 4) in diameter but the uterovesical space was narrow. The bilateral fallopian tubes were infantile and lacked fimbrial ends (Fig. 5). Detorsion of the right ovary was performed.
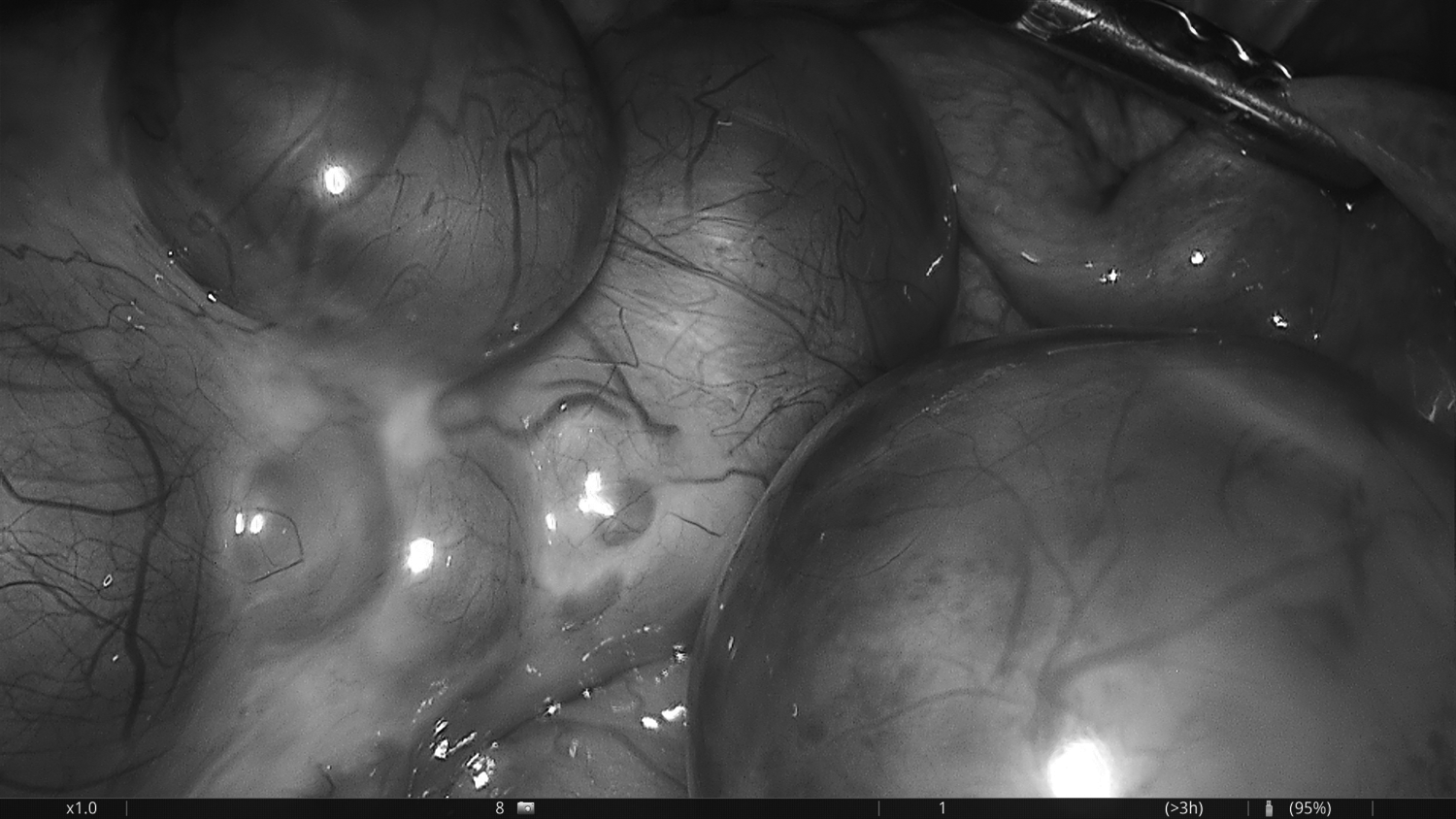
Enlarged and edematous 720° rotated right ovary.
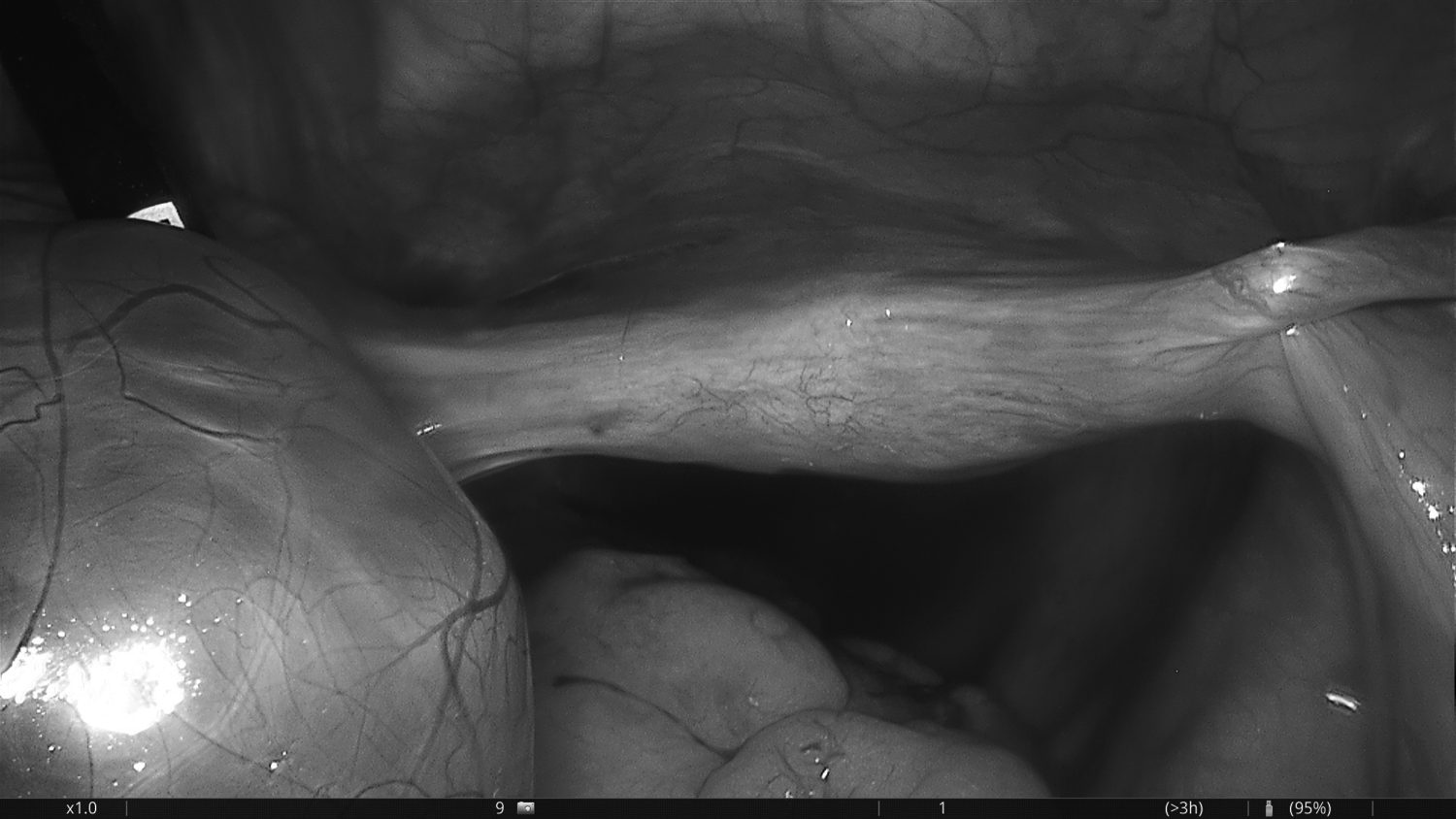
Hypoplastic uterus.
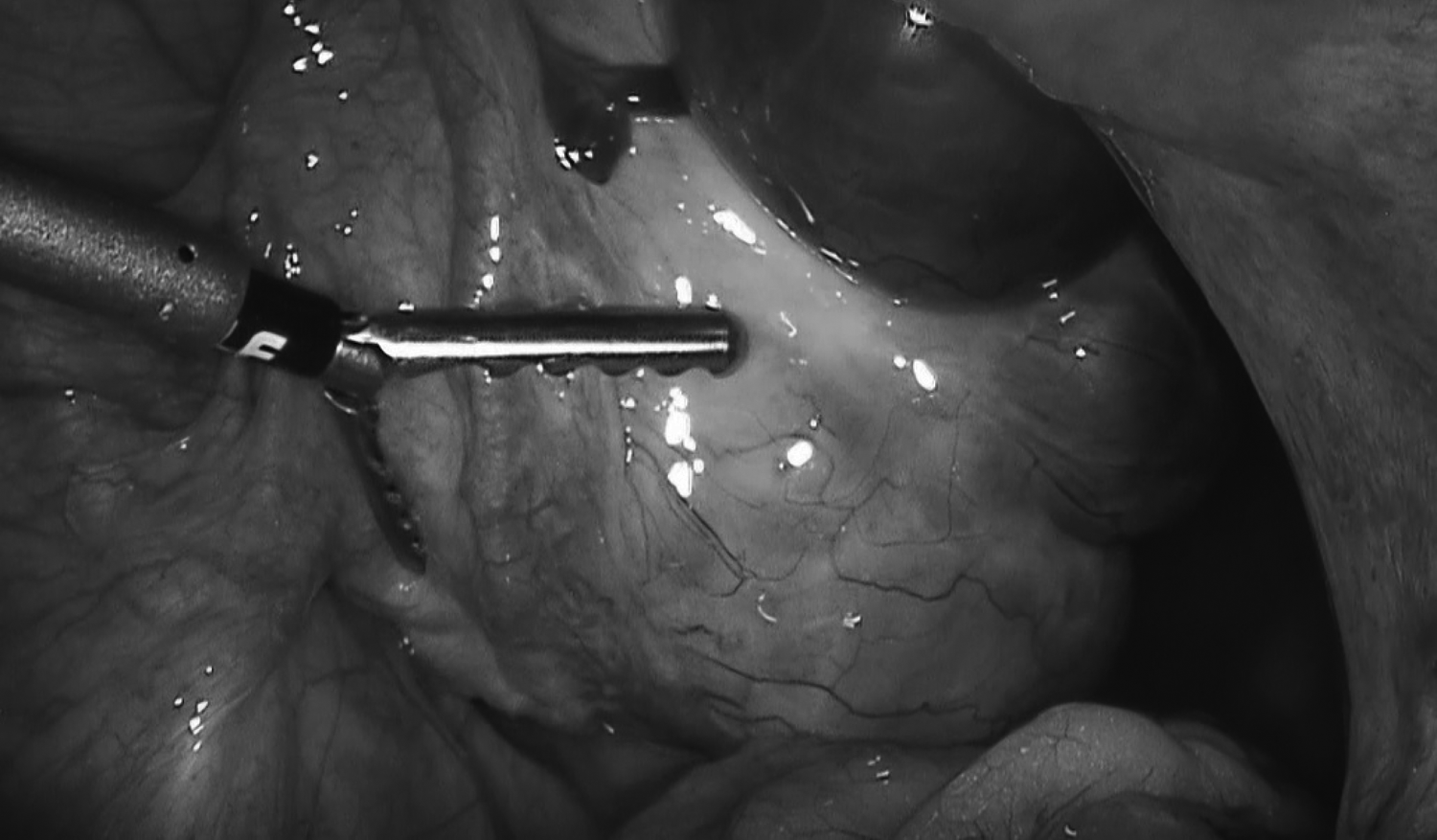
Fallopian tube without fimbrial endings.
Results
The patient recovered well from her laparoscopic surgery.
Laboratory investigations revealed that she had high follicle-stimulating hormone (FSH; 31.28 mIU/mL; normal range: 1.7–7.7 mIU/mL), high luteinizing hormone (LH; 32.35 mIU/mL; normal range: 1–11.4 mIU/mL), low estradiol (E2; < 5 pg/mL; normal range: 22.3–341 pg/mL), normal prolactin (24.1 ng/mL; normal range: 1.9–25 ng/mL), normal total testosterone (0.5 ng/mL; normal range: 0.1–0.5 ng/mL), normal thyroid stimulating hormone (2.2 μU/mL; normal range: 0.53–3.59 μU/mL), normal CA-125 (14.8 U/mL; normal: <35 U/mL), and normal 17-OH progesterone (2.41 ng/mL; normal range: 0.2–3 ng/mL). Fasting glucose, renal and hepatic function tests also showed normal levels.
Her mother has a deeper voice, and when mother was asked for details about her own medical issues, she reported that she had developed virile characteristics—such as the deeper voice, a masculine distribution of hair on her body and pubis, growth of her clitoris, and alopecia during her pregnancies. Combination therapy (0.035 mg of estrogen and 2 mg of cyproterone acetate) was initiated, and she asked for further evaluation after this therapy. Although the patient and her family were well-informed, they did not come back for further evaluations. The patient has 1 sister and 1 brother. The patient's sister had been admitted to another university hospital at age 14 with the same symptoms 1 year prior. The patient's sister underwent surgery and was diagnosed as having a cyst of the corpus luteum. She had partial labial fusion and clitoromegaly with Tanner stage 3 pubertal development. She was 46,XX according to genetic counseling. The diagnosis of congenital adrenal hyperplasia was ruled out. However, her family did not accept further evaluations and she was discharged from hospital at their own request.
Discussion
Placental aromatization of androgens protects both the female fetus and mother against the virilizing effects of fetal androgens. There are several activities of enzymes in different tissues, influencing sexual differentiation, patterns of gonadotropin secretion by the hypothalamic–pituitary axis, reproduction capacity, lipid metabolism, insulin sensitivity, and skeletal maturation and growth. Enzyme catalyzes the synthesis of estrogens from androgens. The aromatase complex is expressed in the endoplasmic reticulum of the cells and consists of two components: (1) cytochrome P450aromatase (cP450arom) and a ubiquitous flavoprotein; and (2) reduced nicotinamide adenine dinucleotide phosphate–cytochrome P450 reductase. Androstenedione, testosterone, and 16 OH-dehydroepiandrosterone sulfate ( 16 OH-DHEAS) arising from fetal liver hydroxylation of fetal adrenal DHEAS are the most common and the most physiologically important substrates for placental estriol synthesis during pregnancy, both in humans and higher primates.
Several molecular CYP19 gene alterations associated with cP450arom deficiency have been described. cP450arom appears to be the product of a single gene located on chromosome 15q21.1. The protein-coding sequence is contained within 9 exons and encodes a protein of 503 amino acids. 18 The core region, defined by a 3-dimensional model of cP450arom, consists essentially of a 4-helix bundle, 2 sheets, and the heme-binding region. The enzyme is expressed in a number of tissues, including the syncytiotrophoblast layer of the placenta; gonads; adipose tissue; bone; brain (hypothalamus, hippocampus, and amygdala); coronary arteries; and various fetal tissues, such as liver, skin, intestine, testis, and ovary. 19
In female newborns with aromatase deficiency, ambiguous genitalia with various degrees of masculinization of the external genitalia have been reported. Gonads were nonpalpable and female internal genitalia differentiation was not affected. In addition, signs of maternal virilization (acne, deep voice, clitoris enlargement) during pregnancy might occur. In a newborn with 46,XX ambiguous genitalia, congenital adrenal hyperplasia should be ruled out first, because of its high frequency and it being a life-threatening condition. Virilizing of a mother during pregnancy is clinically important, because it is not seen in congenital adrenal hyperplasia although it occurs often with aromatase deficiency.
In female infancy and childhood, ovarian cyst development; effect on growth, skeletal maturation, and bone homeostasis; and changes in insulin sensitivity and lipid profile can be seen in affected girls. Basal levels of serum androgens remained high and multiple follicular cysts developed in both ovaries in the current patient. It is known that chronic exposure to abnormally high levels of LH stimulates ovarian cyst development. Androgens, such as those present in polycystic ovary syndrome, promote follicular growth by amplifying the FSH effect. It is accepted that estrogens are important for preserving bone mineral density; a delay in bone maturation is also present in aromatase deficiency.
In female puberty, if complete aromatase deficiency exists, an affected female shows absence of the onset of puberty with a lack of breast development and primary amenorrhea as well as excessive virilization. The pubertal onset is absent and bone aging is delayed. Basal LH and FSH levels are elevated. Serum androgens (testosterone and androstenedione) levels are high, whereas serum E2 levels are extremely low. Insulin resistance and glucose intolerance are associated with high levels of androgens. Multiple ovarian cysts are also seen on pelvic US. During puberty and adulthood, daily estrogen and progesterone therapy is needed for breast development, maintenance of menses, bone mineralization and fusing of epiphysis, and decreasing of gonadotropins.
Serum levels of total testosterone in the current patient were at the upper limit of normal values. This situation was explained by the current authors according to differences of detecting true testosterone levels in the laboratory. The current authors also thought that this patient had partial aromatase deficiency according to the reports from various clinical specialists. Her breast development and menstruation periods were due to low levels of residual aromatase activity.
The term cytochrome P450 oxidoreductase deficiency (POR deficiency) describes a group of allelic conditions characterized by disordered steroidogenesis. Affected enzymes are 21 hydroxylase, 17-20 lyase, and aromatase. Individuals with ambiguous genitalia and/or milder skeletal anomalies appear to fall somewhere in between the mild and severe end of the POR deficiency spectrum. ABS is a severe form of deficiency characterized by several severe congenital craniofacial and skeletal anomalies such as: craniosynostosis; brachycephaly; severe midface hypoplasia; radiohumeral synostosis; and multiple joint contractures. Laboratory tests show that pregnenolone, progesterone, 17-OH pregnenolone, and 17-OH progesterone serum concentrations are often elevated at baseline.
Conclusions
In the etiology of 46,XX disorders of sexual development, aromatase deficiency should be considered after ruling out congenital adrenal hyperplasia. However, detecting signs of virilization in the mother is useful clinical information for diagnosing aromatase deficiency, as it does not occur in congenital adrenal hyperplasia. Pregnenolone, progesterone, 17-OH pregnenolone, and 17-OH progesterone serum concentrations are often elevated in POR deficiency. The current patient was diagnosed as having aromatase deficiency with these findings, but the current authors could not prove this with genetic evaluation because of the patient's nonattendance for a genetic follow-up.
Footnotes
Author Disclosure Statement
No financial conflicts exist.