
Abstract
Abstract
Background:
Leiomyomas arising from Müllerian remnants are a rare association with few case reports in literature. Most of the case reports have shown management with laparotomy. This report presents a case of a leiomyoma arising in a Müllerian remnant that was managed with laparoscopy.
Case:
A 33-year-old nulligravida presented with dyspareunia since marriage and an increase in urinary frequency since 3 years prior to presentation with no history of cyclical abdominal pain. Pelvic examination revealed a vaginal length of 7 cm and a 6 × 6 cm, firm right adnexal mass. Magnetic resonance imaging was suggestive of a hypoplastic uterus with a submucosal myoma or a broad ligament myoma. Laparoscopy showed a bilobed leiomyoma arising from the right Müllerian anlage with the right fallopian tube and ovary attached to its postero-superior surface. The bilobed leiomyoma, and the right Müllerian remnant along with the left rudimentary horn and both tubes were removed as a single contiguous specimen.
Results:
The patient's postoperative recovery was uneventful. Histopathology results were suggestive of a benign leiomyoma. On follow-up at 3 months, this patient was symptomatically improved.
Conclusions:
Leiomyomas from Müllerian remnants are rarely associated, which should be kept in mind in women with Mayer–Rokitansky–Küster–Hauser syndrome presenting with a pelvic mass. Laparoscopic management of such cases is feasible with the added advantages of minimally invasive surgery. (J GYNECOL SURG 33:276)
Introduction
M
Case
A 33-year-old nulligravida female, who was married for 11 years, presented with a history of primary amenorrhea with dyspareunia since her marriage, an increase in urinary frequency since 3 years prior to presentation, and occasional lower backaches. There was no history of cyclical or lower abdominal pain. On examination, it was noted that her secondary sexual characteristics were well-developed. Local examination revealed normal external genitalia and a normal external urethral orifice. On gynecologic examination, her vaginal length was noted to be 7 cm, with a 1 × 1 cm rudimentary uterine nodule at the vaginal apex; a 6 × 6 cm adnexal mass was felt in the right fornix, which was firm in consistency with restricted mobility.
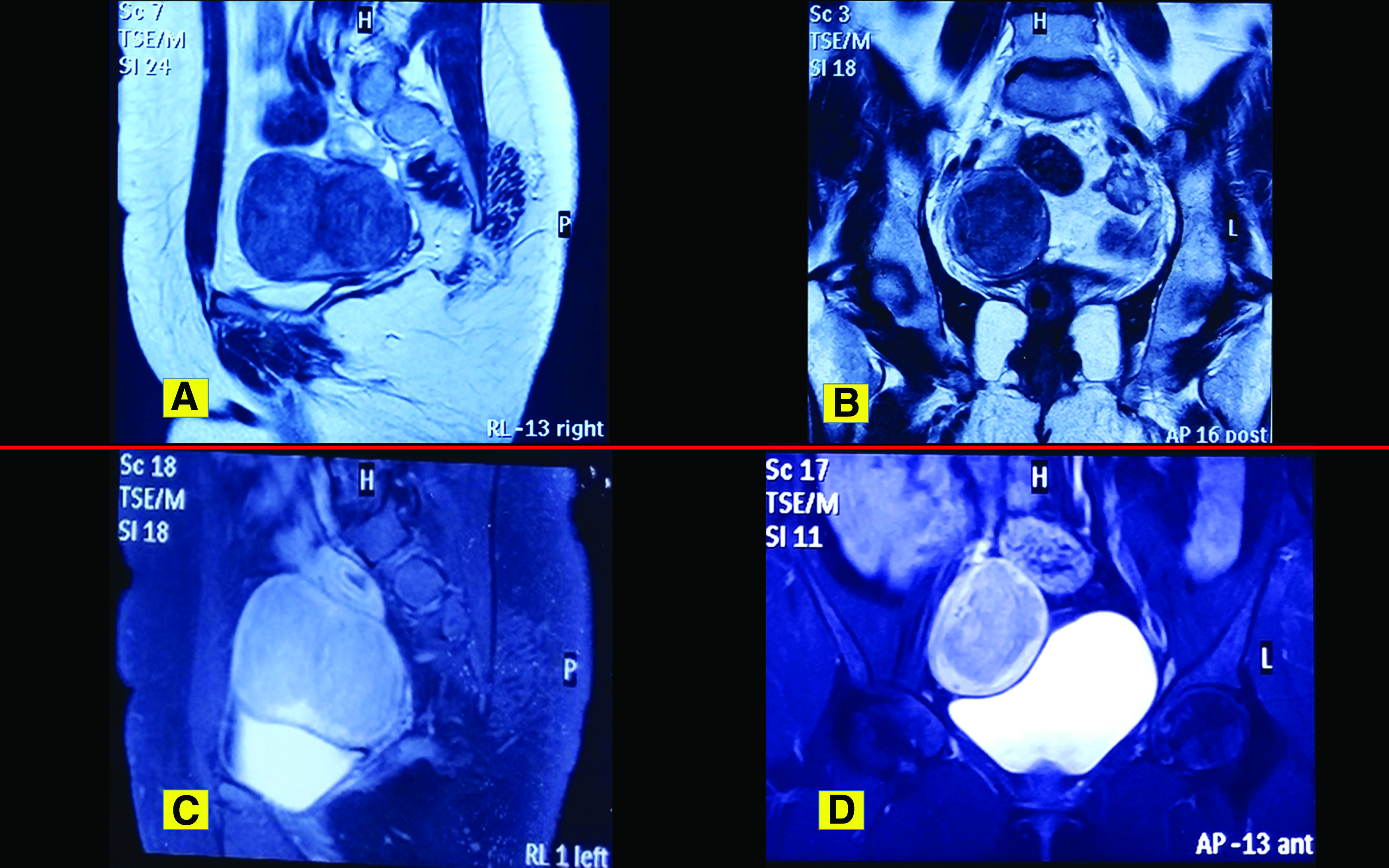
Ultrasound testing was suggestive of a hypoplastic uterus with a broad ligament myoma. Magnetic resonance imaging (MRI) of the pelvis revealed a hypoplastic uterus of ∼1.9 × 1.5 cm, with an absent cervix. MRI also revealed an upper vaginal canal with a complex right adnexal mass of 7.6 × 5.3 cm that was a well-defined bilobed mass, with a peripheral thick rim of T1 hypointense, a T2 hyperintense signal, and a central T1/T2 hypointense signal occupying the right adnexa (Fig. 1). Only the lower one-third of the vagina was visualized. The right ovary was seen separately at the superior and posterior margins of the bilobed mass. Post-contrast images showed intense enhancement throughout this mass. A provisional diagnosis of a hypoplastic uterus with a submucosal fibroid or a broad ligament fibroid with degeneration was made. An intravenous pyelogram was performed, which was suggestive of normally positioned bilateral kidneys and ureters. Her cancer antigen–125 and germ-cell tumor marker levels were within normal limits. Her karyotype was 46XX. Her hormonal profile was within normal limits. Her urine culture was sterile.
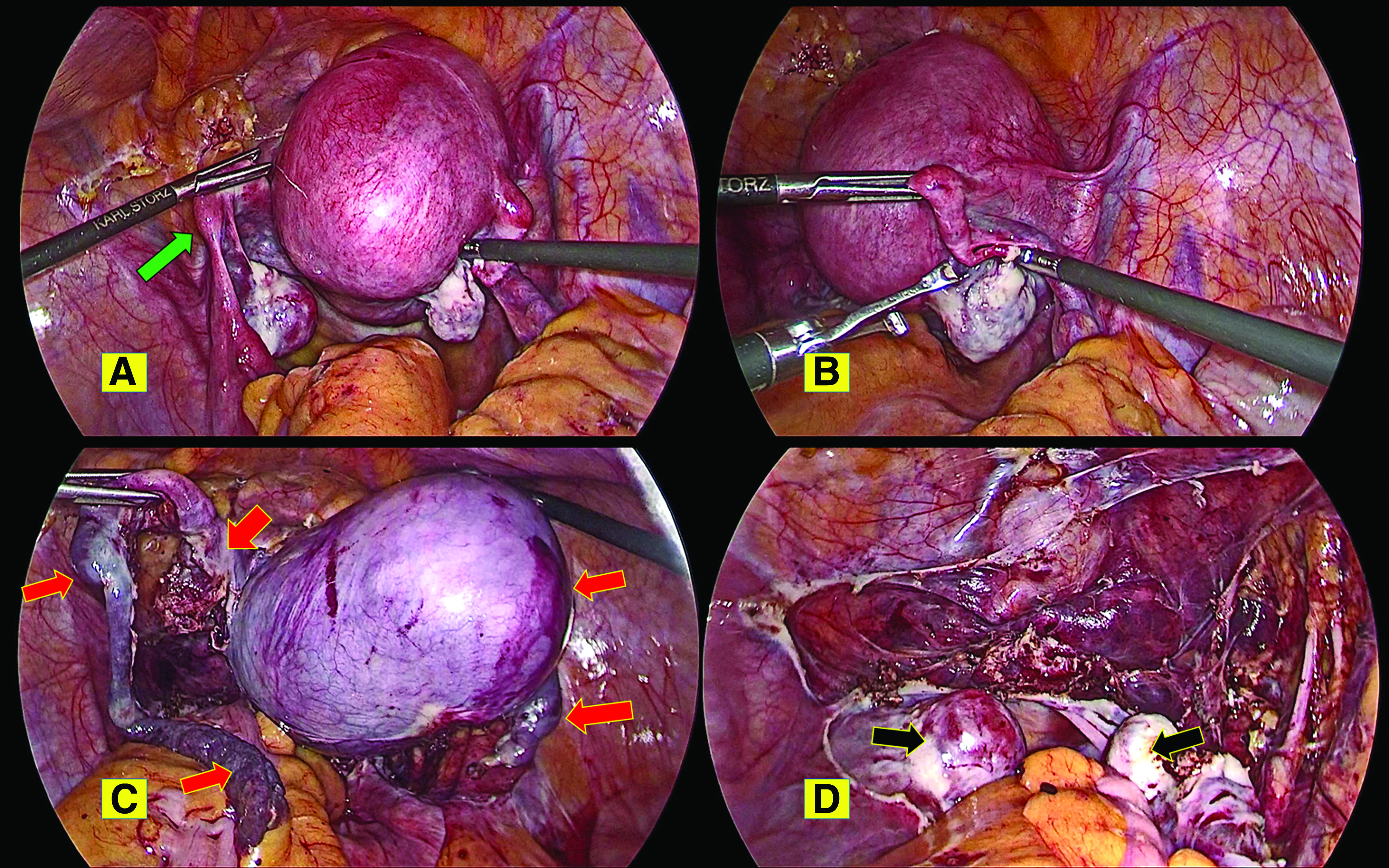
With a provisional diagnosis of a pelvic leiomyoma, this patient was scheduled for laparoscopy and this was performed. Intraoperatively, the mass was noted to be a bilobed leiomyoma arising from the right Müllerian-duct remnant, with the right fallopian tube and ovary attached to its posterior superior surface (Fig. 2). A fibrous band was present connecting the right bilobed leiomyoma and left rudimentary horn, with its attached left fallopian tube and ovary. After dissection by harmonic scalpel and a bipolar device, the bilobed leiomyoma along with the left rudimentary horn, fibrous band with a rudimentary nodule, and both fallopian tubes were removed as a single contiguous specimen. Contained-in-bag mechanical power morcellation was performed and the specimen sent for histopathology testing. The bilateral ureteric course was traced and the bladder integrity was checked by retrograde filling with saline and methylene blue dye. The operation time was 90 minutes. The patient's blood loss was ∼50 mL.
Results
The patient's postoperative recovery was uneventful. Due to having an adequate-sized vagina, she was advised to resume regular intercourse after 2 weeks. At her 3-month follow-up, she was symptomatically improved.
Histopathology results were suggestive of a benign leiomyoma. The left rudimentary horn had endomyometrial fragments, and the bilateral fallopian tubes were within normal histologic limits.
Discussion
MRKH syndrome is the second most common cause of amenorrhea. The karyotype is 46XX. The main differential diagnosis is androgen insensitivity syndrome, which is a 46XY karyotype with an absence of all Müllerian structures and intra or extra-abdominal gonad (testis) location. Once MRKH is clinically suspected, imaging studies, such as ultrasounds and MRI, helps determine the degree of abnormalities, with MRI showing an overall correlation of >95% between MRI and laparoscopic findings. 2 Sometimes, if MRI fails to demarcate either exact uterine abnormality or precise anatomical location of tubal remnants and ovaries, then laparoscopy can be used for making a diagnosis. 3 A probable explanation for findings seen in women with MRKH would be either an abnormal activation of Müllerian-inhibiting substance or altered expression and potential dysregulation of two groups of genes. These genes are, namely, HOXA and WNT4, which are implicated in normal development of Müllerian, renal, and bone structures.2,4 MRKH syndrome is of two types. Type I, or the isolated or typical form or Rokitansky sequence, is characterized by the presence of symmetrical muscular buds or Müllerian remnants with normal fallopian tubes, where only the caudal part of the paramesonephric ducts is affected. 5 Type II, or the atypical form, is characterized by asymmetrical hypoplasia of one or two muscular buds, with or without dysplastic fallopian tubes, and is often associated with abnormalities of additional organ systems, such as the renal system or skeletal system. 6 The most severe atypical form of the disorder is termed MURCS [Müllerian Renal Cervical Somite] association, which is characterized by Müllerian duct (MU) aplasia, renal (R) aplasia, and cervicothoracic somite (CS) dysplasia. 7
The present case was type I MRKH syndrome because of the presence of bilateral Müllerian anlages with leiomyoma development in the right anlage. Leiomyomas are benign smooth-muscle–cell tumors that can arise from any organ containing smooth-muscle cells. Leiomyomas of the uterus are the most common benign uterine tumors and benign pathologies of the female genital tract. Their incidence increases with age, and a study from the United States has shown a 51% incidence of fibroids noted on ultrasounds in premenopausal women with no previous diagnoses. 8 An 8-year follow-up study to determine the risk of having a major uterine procedure have shown the presence of fibroids in 64% of women between ages 35 and 49 in baseline scans. 9 Examination of hysterectomy specimens has shown a 77% fibroid prevalence. 10
Due to the presence of smooth-muscle cells in the proximal part of the Müllerian ducts, theoretically, leiomyomas are possible in the Müllerian-duct remnants in women with MRKH syndrome. 11 Similar to leiomyomas in a normal uterus, spontaneous chromosome rearrangements could be responsible for initiation and progression of leiomyoma growth in Müllerian remnants. 12 Leiomyomas in cases of Müllerian agenesis or dysgenesis presenting as pelvic masses are rare occurrences, with few case reports in literature.13–20 Most of the case reports have shown similar occurrences of leiomyomas in Müllerian-duct remnants in women between ages 30 and 45. This age range is also the most common period in a women's life for the development of leiomyomas in normal myometrium. Few case reports of adenomyosis in the rudimentary horn in patients with MRKH syndrome were also described.21,22
A patient with MRKH syndrome who has an adnexal mass poses a diagnostic dilemma for a clinician. Vaginal agenesis with a pelvic mass can be a hematometra or a hematosalpinx, but the absence of cyclical abdominal pain ruled out the presence of functional endometrium in the current case. This patient had urinary frequency for 3 years prior to presentation, probably due to the pressure effect of the leiomyoma. Other causes of pelvic pathology—such as renal anomalies, dysgenetic gonads with gonadal tumors, ovarian pathology, retroperitoneal tumors, and bladder tumors—should be ruled out before proceeding to management of these patients. For differential diagnosis, a clinician may consider computed tomography or MRI; karyotyping; and testing for tumor-marker levels to derive a possible diagnosis. Definitive diagnosis and treatment with laparotomy or laparoscopy is necessary as a part of the management of such patients. Most of the case reports were about managing the patients with laparotomy. The first case managed with laparoscopy was reported in 2000 by Tsin et al. 23 The current case report supports laparoscopic diagnosis and management in such patients.
Women with MRKH often face psychologic and social issues due to sexual insecurity and inability to bear children in their wombs. 2 When addressing the sexual insecurity, the goal is to construct a canal between the bladder and rectum with a proper moist lining to permit sexual intercourse with vaginal dilation therapy being the first-line, safe and preferred method. 24 For absolute uterine-factor infertility, these women can opt for adoption, or gestational surrogacy, or for enjoying biologic and gestational motherhood, a human uterine transplantation is an emerging option. 25 In the present case, the woman had a 7-cm vagina, which developed with regular sexual intercourse, so she was advised to continue with the same intercourse and also to opt for surrogacy in the follow-up.
Conclusions
Leiomyomas arising from Müllerian-duct remnants are a rare association. These tumors should be kept as a differential diagnosis in a case of solid pelvic tumors in women with MRKH. Laparoscopic diagnosis and management of such cases is feasible, with the added advantages of minimally invasive surgery.
Footnotes
Acknowledgments
The authors would like to thank the patient and her husband for their consent to publish this case report.
Author Disclosure Statement
None of the authors has any financial conflicts to disclose.