
Abstract
Background:
Ovotesticular disorder of sexual development (OT-DSD) is a rare condition characterized by abnormal gonadal development. Although most OT-DSD is diagnosed during infancy, diagnosis is often delayed or never made in countries with limited resources.
Case:
A 21-year-old Haitian male presented with gynandromorphic genitalia and breast development, and desired surgical correction of his feminized anatomic features. Physical examination revealed bilateral symmetrical breasts. External genitalia revealed a 7 cm penile-like structure adherent to the ventral aspect of the clitoral hood. A small presumed testis within the scrotum was noted on the right side, whereas the testis and scrotum were absent on the left. Computer tomography of the abdomen and pelvis demonstrated normal kidneys, bilateral ureters, a uterine horn, an intra-abdominal gonad on the left, a descended gonad on the right, and a structure consistent with a vagina.
Results:
He underwent gender affirmation surgery in two stages: first, an abdominal hysterectomy, left salpingo-oophorectomy, right trans-scrotal testicular biopsies, and bilateral simple mastectomy, then penile reconstruction and metoidioplasty. The right trans-scrotal biopsy confirmed testicular tissue and left-sided gonad was consistent with ovarian tissue. Traditional cytogenetic analysis revealed 46, XX karyotype. His postoperative course was uncomplicated and 6-month follow-up demonstrated an excellent surgical outcome.
Conclusion:
Gender affirmation surgery can improve the quality of life of patients who are diagnosed with OT-DSD after puberty. However, access to care continues to be a problem in resource-poor settings and is a major limitation in the treatment.
Background
Ovotesticular disorder of sexual development (OT-DSD), previously known as true hermaphroditism, is a rare condition that is characterized by the presence of both testicular and ovarian tissue within an individual. 1 The incidence varies geographically. In North America and Western Europe, the incidence of OT-DSD is <10% of all DSD disorders, whereas in South Africa the prevalence is >51%.2,3 In majority of cases, the diagnosis of OT-DSD is ascertained at the time of newborn evaluation. Unfortunately, in undeveloped countries, identification and treatment of OT-DSD can be delayed due to insufficient resources. Subsequently, during puberty, these individuals face the challenge of defining their gender and sexuality. Sexual ambiguity can have a significant impact on psychologic, social, and emotional well-being, and additionally presents a therapeutic challenge for the treating physician. We present the case of a 21-year-old who identified as male with gynandromorphic genitalia and breast development. We discuss the diagnostic approach and treatment of OT-DSD in a resource-poor setting, and the challenges faced by the medical team who participated in their care.
Case
A 21-year-old male with no significant past medical history presented to a clinic in Port-au-Prince, Haiti, with complaints of progressively enlarging breasts since the age of 15. He denied having menstruation, however reported abdominal cramping every month beginning at the age of 16. He reported being sexually active with the ability to have erections, but was unable to produce semen.
On physical examination, Tanner Stage V breast development was noted with normal distribution of both facial, axillary, and pubic hair. Examination of the external genitalia revealed a 7 cm long penile-like structure that was immobile and adherent to the ventral aspect of a clitoral hood (Fig. 1A). A small gonad was noted within the right labioscrotal sac; however, no gonadal structure was identified on the left. A small opening on the perineum, underneath the penile-like structure was noted (Fig. 1A, B). This was later identified as the urethral meatus during examination under anesthesia. Computer tomography of the abdomen and pelvis was obtained. The study demonstrated normal kidneys, bilateral ureters, a uterine horn, an intra-abdominal gonad on the left, a descended gonad on the right, and a structure consistent with a vagina. Unfortunately, due to limited resources chromosomal analysis could not be obtained and further evaluation and treatment was delayed.

The findings on examination and imaging were reviewed with the patient. With consideration of the patient's wishes, several options were discussed. The patient was counseled on options for treatment, including observation, removal of the intra-abdominal gonad and female reproductive organs only, gender affirming surgery as female or, gender affirming surgery as male. The risk and benefits of orchiectomy was also discussed. The risk of developing testicular cancer in the future was weighed against lack of access to hormone replacement therapy. After extensive counseling and discussion, the patient opted for gender affirmation surgery as a male and to forego testicular removal.
Surgery was performed in a staged manner. At the initial surgery, intraoperative findings noted a small boggy uterine horn, a left normal-appearing ovary, and a dilated left fallopian tube. Bilateral ureters were identified in the normal anatomical position leading to a normal appearing bladder. An abdominal hysterectomy, left salpingo-oophorectomy, right central and peripheral trans-scrotal testicular biopsies, and bilateral simple mastectomies were performed. The right testicular gonad was left in situ.
Results
All the specimens obtained and blood work performed at the time of the initial surgery were transported to the United States in containers at temperatures recommended by our cytogenetics laboratory. Traditional cytogenetic analysis and fluorescence in situ hybridization on the male determining region of the Y chromosome (SRY gene) was performed. Results revealed 46, XX karyotype with negative translocation of the SRY gene, which is consistent with a female karyotype. Several hormonal tests were also performed to further evaluate the hypothalamic-pituitary-gonadal axis and can be seen in Table 1. The results affirmed female hormonal profile in the follicular phase of the menstrual cycle and ruled out congenital adrenal hyperplasia.
Laboratory Testing Obtained for Diagnosis of Ovotesticular Disorder of Sexual Development
F, follicular; L, luteal.
Histopathologic examination of the central trans-scrotal biopsy confirmed testicular tissue, which demonstrated atrophic seminiferous tubules with focal hyalinization. Spermatogenesis was absent. Leydig cells were present in small groups around the seminiferous tubules. The peripheral trans-scrotal biopsy demonstrated a fragment of epididymis with no significant pathologic findings. Examination of the left-sided gonad was consistent with an ovary, which demonstrated the presence of multiple corpora lutea in varying stages of involution, corpora albicantia, and primary follicles in various stages of development. Sections of the fallopian tube showed an ectatic duct with hemorrhagic content, lined by an unremarkable ciliated serous epithelium. There was no evidence of endometriosis. The uterus was lined with mid-secretory endometrial glands with intraluminal secretions and stromal edema.
The second stage of the surgery took place 6 months after the initial surgery. He underwent gender affirming surgery that included penile reconstructive surgery and metoidioplasty. This was performed by dividing the skin of the phallus/clitoris from the underlying cavernous body. By dissecting the cordee that was tethering the phallus ventrally, the straightening of the cavernous body was achieved. Reconstruction of the urethra was accomplished by advancing the meatus from the base toward the tip of the phallus. The dorsal position of the urethra was reconstructed using a graft of buccal mucosa, whereas the ventral portion was created with a flap of skin mobilized from the right labia minora. The reconstruction of the penis was completed by advancing another skin flap from the left labia majora (Fig. 2A, B). The distance between the native meatus and the tip of the phallus was 10 cm at the conclusion of the procedure.
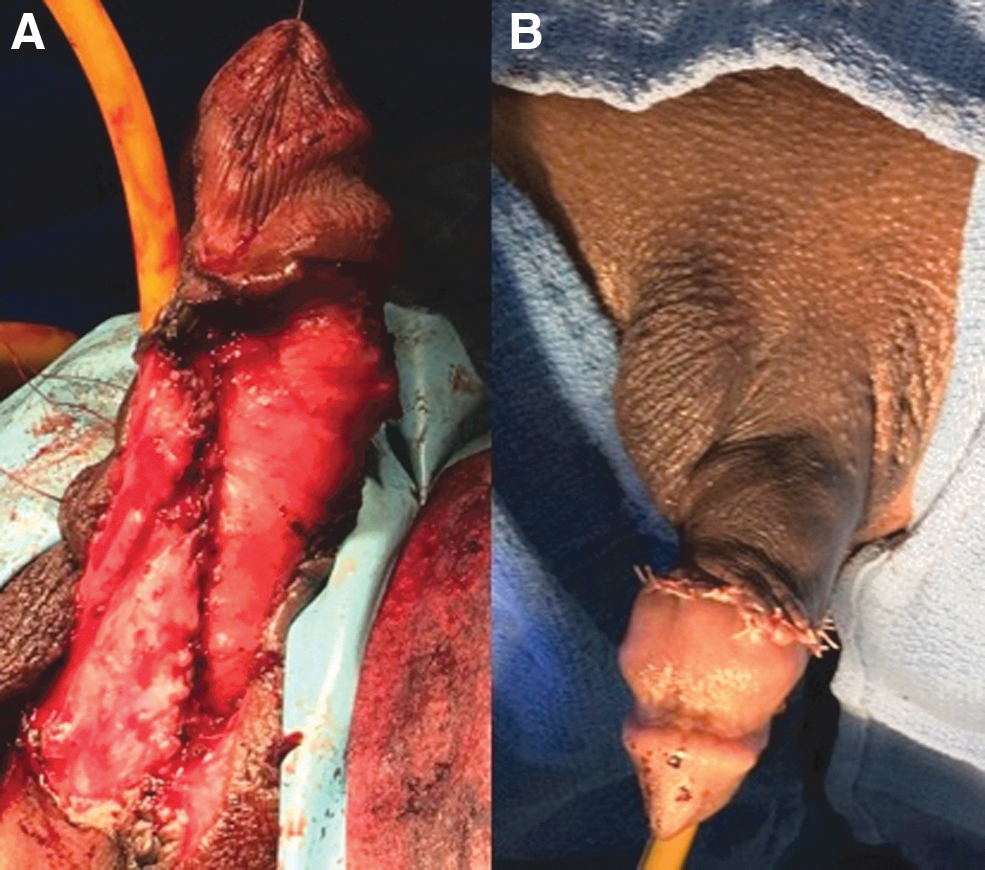
At the patient's 6-month follow-up, he demonstrated an uncomplicated postoperative course and excellent surgical outcome as the patient reported a satisfactory level of sexual function.
Discussion
OT-DSD is a rare disorder of abnormal gonadal development defined as an individual who has congenital presence of both ovarian and testicular tissue. 1 The most prevalent karyotype in OT-DSD is 46, XX, comprising >80% of cases. Less commonly observed karyotypes are 46, XY, 46, XX/46, XY, and 46, XX/47, XXY due to mosaicism or chimerism.1,2,4 The genetic mechanisms and pathophysiology of OT-DSD has yet to be fully understood. Currently, a translocation involving the SRY gene or mutations that promote testicular development are thought to be contributory to the process of how testicular development occurs in OT-DSD. 1 In a case report of 33 patients with OT-DSD, only 35% of cases with 46, XX karyotype was noted to have positive SRY gene. 4 Our patient was found to be SRY negative.
The most commonly identified gonad in OT-DSD is an ovotestis, followed by ovary then testis. 2 Gonads containing testicular tissue are more commonly seen on the right, whereas gonads containing ovarian tissue are seen frequently on the left, 1 which is consistent with the findings in our patient. The internal structures typically reflect the ipsilateral gonad differentiation. 1 Testes and ovotestis are commonly atrophic with hyalinization and absence of spermatogenesis. The ovaries are often functional, leading to the development of breasts in ∼75% of those with OT-DSD and menstruation in ∼50%.1,2 External genital development reflects the level of androgen production and exposure. As a result, phenotypes are highly variable. 1
Inspection of external genitalia is a routine part of the standard newborn physical examination. In cases of ambiguous genitalia, an extensive evaluation is initiated to determine the newborn's karyotype and the etiology of ambiguity. Although most cases of OT-DSD are diagnosed during infancy, rarely diagnosis can occur late, particularly in developing countries.5,6 Delayed diagnosis of OT-DSD harbors not only economical and health care challenges, but is also paired with sociocultural, psychologic, and emotional complexities. 7 Patients diagnosed with ambiguous genitalia may be ostracized in their own community due to a lack of knowledge and prejudicial cultural issues. Unfortunately, our patient was no exception and endured significant discrimination from his community.
Currently, there is continued debate regarding timing of gender affirmation surgery for those with OT-DSD. Although some advocate initiating gender assignment early in childhood, others prefer to reserve this option until the individual reaches gender maturity.6–8 In addition, consideration of foregoing orchiectomy to preserve male hormone levels for our patient was essential due to lack of access to hormonal therapy in this resource-poor setting and balancing this with the risk cancer in the future. Although our patient did experience a delayed diagnosis of OT-DSD, he was greatly satisfied with his surgery.
Conclusion
In conclusion, this case demonstrates the continuing struggle that resource-poor settings have in obtaining the health care necessary to diagnose and treat OT-DSD, leaving patients vulnerable to the challenges of gender identity on an individual and social level, and challenges encountered by the managing medical team in streamlining the diagnosis and treatment. In addition, it also highlights the need for increased awareness and training of personnel so that ambiguous genitalia can be identified in timely manner and the necessary referral, evaluation, and treatment can be initiated.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this study.