
Abstract
Abstract
Mariggiò, Maria A., Stefano Falone, Caterina Morabito, Simone Guarnieri, Alessandro Mirabilio, Raffaele Pilla, Tonino Bucciarelli, Vittore Verratti, and Fernanda Amicarelli. Peripheral blood lymphocytes: a model for monitoring physiological adaptation to high altitude. High Alt. Med. Biol. 10:333–342, 2010.—Depending on the absolute altitude and the duration of exposure, a high altitude environment induces various cellular effects that are strictly related to changes in oxidative balance. In this study, we used in vitro isolated peripheral blood lymphocytes as biosensors to test the effect of hypobaric hypoxia on seven climbers by measuring the functional activity of these cells. Our data revealed that a 21-day exposure to high altitude (5000 m) (1) increased intracellular Ca2+ concentration, (2) caused a significant decrease in mitochondrial membrane potential, and (3) despite possible transient increases in intracellular levels of reactive oxygen species, did not significantly change the antioxidant and/or oxidative damage-related status in lymphocytes and serum, assessed by measuring Trolox-equivalent antioxidant capacity, glutathione peroxidase activity, vitamin levels, and oxidatively modified proteins and lipids. Overall, these results suggest that high altitude might cause an impairment in adaptive antioxidant responses. This, in turn, could increase the risk of oxidative-stress-induced cellular damage. In addition, this study corroborates the use of peripheral blood lymphocytes as an easily handled model for monitoring adaptive response to environmental challenge.
Introduction
Paradoxically, exposure to a hypoxic environment is known to increase ROS concentration in various cells and tissues as a result of the activity of complex III of the mitochondrial respiratory chain (Guzy and Schumacker, 2006; Murphy, 2009). Many researchers have reported important redox imbalances after prolonged mountain sojourns (Sinha et al., 2009). Some studies have indicated possible pathogenic roles of oxidatively modified molecules generated in response to hypoxia (Behn et al., 2007). In fact, an overproduction of ROS can lead to the oxidation of essential biomolecules, including lipids, proteins, and nucleic acids, and may be associated with cellular dysfunction, thus promoting various biological responses, such as inflammation and apoptosis (Ames et al., 1993; Breen and Murphy, 1995; Droge and Schipper, 2007; Karihtala and Soini, 2007).
Intense physical activity is also known to promote ROS overproduction, mainly through oxygen overconsumption, ischemia–reperfusion-induced activation of xanthine oxidase, neutrophil-mediated superoxide release resulting from tissue damage, and inflammatory processes (Fisher-Wellman and Bloomer, 2009). Therefore, high-energy-demanding mountain expeditions may aggravate oxidative damage that could result from exposure to low oxygen tension, thus exacerbating the risk of developing oxidative-stress-induced dysfunction and pathologies. On the other hand, exercise-induced increases in the generation of ROS also trigger adaptation mechanisms that may decrease the incidence of ROS-associated diseases (Radak et al., 2001).
As reported by several authors, a close mutual relationship exists between increased ROS production and intracellular Ca2+ concentration ([Ca2+]i). In fact, modification of cytoplasmic Ca2+ levels could be one of the most important mechanisms by which ROS exert their multiple actions in cells (Camello-Almaraz et al., 2006; Feissner et al., 2009). Several Ca2+ transport systems are modulated by oxidation. For example, the activity of inositol 1,4,5-trisphosphate and ryanodine receptors, the main Ca2+ channels of intracellular stores, depends on oxidation status, which thus regulates spontaneous and stimulus-induced intracellular Ca2+ oscillations (Davidson and Duchen, 2006). Mitochondria, the main ROS producers, are also known to control intracellular Ca2+ signals through Ca2+ uptake and release during cytosolic Ca2+ mobilization (Nicholls, 2005). Furthermore, there is evidence that Ca2+-mobilizing stimuli generate mitochondrial ROS, which, in turn, could facilitate intracellular Ca2+ signaling (Camello et al., 2000; Xi et al., 2005).
Extensive research has been dedicated to the discovery of unique and easily available biological models for integrated studies concerning physiological and pathological conditions in relation to whole-body exposure to environmental stressors. Defining the role of environmental changes in the control and alteration of the oxidative balance is limited by two main factors: (1) our understanding of the persistence of environmentally induced oxidative alterations and (2) the availability of a cellular system easily obtained from a single source, yet capable of providing valuable information on the status of the entire body. The invasiveness of the procedure required to obtain adequate answers to the questions raised is an additional factor that needs to be taken into account.
As previously reported, peripheral lymphocytes can be a reliable model for studying the pathophysiology of oxidative-stress-mediated Ca2+ homeostasis alterations, which induce cell dysfunction and lead to organic pathogenic states (Belia et al., 2009). In addition, the hematopoietic system is highly sensitive to environmental factors, and the blood system is known to be directly involved in the development of tolerance to hypoxic conditions (Chouker et al., 2005; Shtemberg et al., 2007; Brooks et al., 2009). Furthermore, because circulating lymphocytes are readily accessible and retain their original phenotype during in vitro culture, they offer important advantages for cellular and molecular studies (Nagaeva et al., 2002).
For all these reasons, we used lymphocytes, which are a “time-persistent” system (Balakrishnan and Rao, 1999; Gregoire et al., 2006) capable of reflecting the condition of the whole organism, as a model to analyze the effects of high altitude exposure. In a longitudinal study, we analyzed oxidative status-related parameters in lymphocyte samples collected prior to, soon after, and 6 months after a high altitude expedition; we also assessed cell Ca2+ balance and mitochondrial function, because these two mechanisms are strictly related to oxidative status.
Our overall goal was to determine if high altitude induces oxidative stress and dysfunction by utilizing peripheral blood lymphocytes as biosensors of the whole-body response and as a model for adaptation of the organism to environmental modifications.
Materials and Methods
Subjects and the expedition
Seven healthy, male, nonsmoking climbers were enrolled in the study; their characteristics are summarized in Fig. 1. The health conditions of climbers were assessed at the time of each blood sampling. Written consent was obtained from each participant, and the study was designed in accordance with the recommendations of the Declaration of Helsinki and approved by the Ethics Committee of the Gabriele d'Annunzio University of Chieti-Pescara, Italy.
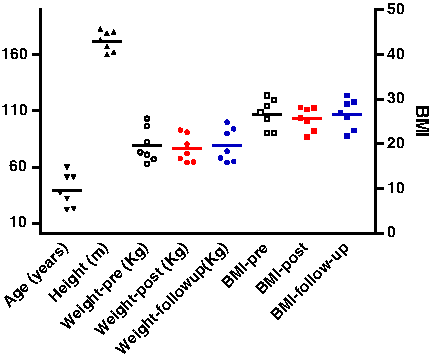
Anthropometric characteristics of the seven subjects. The values of age, height, and weight are plotted on the left y-axis, while the values of BMI are on the right y-axis.
The climbers took part in the INTERAMNIA 8000–MANASLU 2008 expedition that started from Rome (sea level: SL) on September 8, 2008, and ended in the same city on October 20, 2008. The subjects had not experienced high altitude conditions within a period of at least 6 months prior to this expedition. The expedition timetable and altitude profile are shown in Fig. 2.
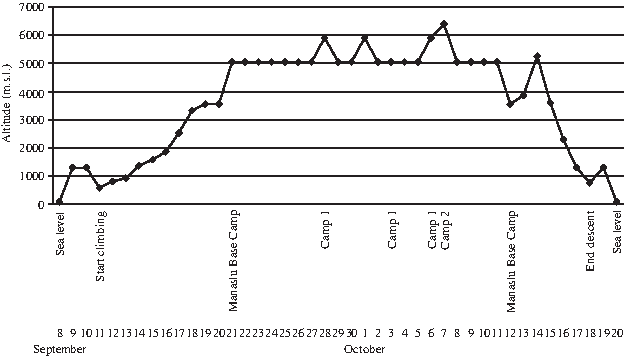
Timetable and altimetric profile of the expedition.
After landing in Kathmandu, Nepal (∼1300 m above SL), on September 9, 2008, climbers began the ascent phase on September 11, gradually reaching the Manaslu Base Camp (5000 m above SL) on September 21. During this 10-day period, climbers covered approximately 25 km/day and averaged about 7 h of trekking each day.
Climbers remained at the base camp for 21 days, during which symptoms of acute mountain sickness were insignificant and decreased after a few days; no climber needed any medication. Throughout the expedition, balanced food intake and ad libitum fluid ingestion were allowed. Antioxidant vitamins were not administered before or during the expedition. When the meteorological conditions were favorable, the climbers reached Camp 1 (5900 m above SL) and Camp 2 (6400 m above SL), covering approximately 8 to 10 km/day in an average 6 h/day of trekking. During the period at the base camp, the climbers busied themselves attending to their personal needs, rearranging tents, and maintaining the camp. Nepalese Sherpa packers and base-camp personnel supported the expedition; however, no data were collected from these Nepalese citizens.
On October 19, the subjects reached Kathmandu and left the following day. After landing in Rome on October 20, climbers were transferred to Chieti, Italy, for health evaluation. Since the logistics and the equipment of the expedition could not ensure safe, controlled, and standardized time-course blood withdrawals, serum isolation, and sample storage, each subject underwent blood withdrawal before, soon after (on October 20, once arrived in Italy), and 6 months after the expedition (follow-up).
Materials
All media, sera, antibiotics, and culture solutions were purchased from Gibco BRL (Paisley, Scotland, UK). All sterile culture plastics were obtained from Falcon (Plymouth, UK). All other reagents were analytical grade.
Isolation of Serum
Sera were isolated from peripheral blood samples, drawn from each subject before and after the expedition, using Terumo test tubes (Venosafe Serum Gel VF-106SAS, Terumo Europe NV Laboratory Systems, Rome, Italy). Blood samples were centrifuged at 3500 rpm for 15 min. Each serum sample was collected and frozen at −80°C prior to biochemical measurements.
Isolation of Human Lymphocytes
Peripheral venous blood samples from climbers were collected in sodium-heparinized vacutainers. Peripheral blood lymphocytes were separated under sterile conditions on a Ficoll-Histopaque 1077 (SIGMA, Milan, Italy) gradient using the Boyum method (Boyum, 1976). Aliquots of heparinized whole blood, diluted with an equal volume of Dulbecco's phosphate-buffered saline (1:1), were gently applied to an equal volume of Ficoll-Paque PLUS in centrifuge tubes. Samples were centrifuged at 400 × g for 30 min, and the resultant interface (buffy coat) was carefully aspirated from the gradient and washed twice in Dulbecco's phosphate-buffered saline by centrifugation at 200 × g for 10 min. The cell pellet was resuspended in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS), 2% (w/v)
Markers of Oxidative Stress
Serum
Total antioxidant status
Because of interactions among antioxidants and difficulties in measuring each antioxidant component separately, methods were developed to assess the total antioxidant status of serum or plasma. For this purpose, we used the 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox)-equivalent antioxidant capacity (TEAC) assay, which is a widely used kit-based commercial method. This assay is based on suppression of the absorbance of radical cations of 2,2′-azino-di-(3-ethylbenzthiazoline sulphonate) (ABTS) by antioxidants in the test sample when ABTS is incubated with a peroxidase (metmyoglobin) and hydrogen peroxide (H2O2) (Rice-Evans and Miller, 1994). Assays were performed as described by the manufacturer of the kit (Cayman Chemical, Ann Arbor, MI, USA). Briefly, 10-μL aliquots of serum was added in duplicate to 10 μL of metmyoglobin and 150 μL of a chromogen solution, and reactions were initiated by the addition of 40 μL of H2O2. Reaction mixtures were incubated for 3 min at room temperature and read using a Victor3 microplate reader (PerkinElmer, Waltham, MA, USA). TEAC values in samples were determined by reference to a linear calibration curve prepared using pure Trolox-containing reactions (range: 0–0.33 mmol/L).
Thiobarbituric acid-reactive substances
The measurement of thiobarbituric acid-reactive substances (TBARS) is a well-established method for detecting lipid peroxidation (Yagi, 1998). We used TBARS Assay Kit, which allows rapid photometric detection at 532 nm of the thiobarbituric acid–malondialdehyde (TBA-MDA) adduct, as described by the manufacturer (cat. 10009055; Cayman Chemical). In brief, 100 μL of serum was added in duplicate to 100 μL of sodium dodecyl sulfate (SDS) and 4 mL of color reagent. Reaction mixtures were then incubated for 1 h in boiling water and centrifuged at 1600 × g for 10 min at 4°C. After warming for 5 min at 25°C, samples were read on a Lambda25 spectrophotometer (PerkinElmer). Values for samples were calculated from a linear calibration curve prepared using pure MDA-containing samples (range: 0–50 μmol/L).
Protein carbonyl content
A colorimetric assay (Protein Carbonyl Assay Kit, cat. 10005020, Cayman Chemical) was used to evaluate oxidized proteins (Levine et al., 1994), as described by the manufacturer. In brief, each serum sample was treated in duplicate with 2,4-dinitrophenylhydrazine (DNPH) dissolved in 2.5 mol/L HCl. The formation of a Schiff base between protein carbonyls and DNPH produced the corresponding hydrazones, which could be isolated from a 20% trichloroacetic acid and ethanol–ethyl acetate (1:1) solution by centrifugation at 10,000 × g for 10 min at 4°C. Hydrazone-containing pellets were redissolved in guanidine hydrochloride and read at 370 nm using a Victor3 microplate reader (PerkinElmer), as described by the manufacturer. The absorbance of samples treated with 2.5 mol/L HCl was subtracted from that of DNPH-treated samples, and the corrected values thus obtained were used to determine the concentration of protein carbonyls (ɛ = 22,000 (mol/L)−1cm−1). Values were then normalized to the total protein concentration in the final pellet (OD280) to take into account protein loss during the washing steps, as suggested in the kit manual.
Vitamin determinations
A high-performance liquid chromatographic (HPLC) method was used to measure vitamins A and E and β-carotene levels in serum using wavelength-programmed, ultraviolet–visible, absorbance detection. After de-proteinization with ethanol, a 200-μL aliquot of plasma from each subject, containing tocopherol acetate as an internal standard, was extracted with butanol–ethyl acetate. Sodium sulfate was added for dehydration. Analytes of extracted samples were found to be stable for at least 4 days. A 10-μL aliquot of each organic extract was used for HPLC analysis. The mobile phase was methanol:butanol:water (89.5:5:5.5, v/v) and the flow rate was 1.5 mL/min. Analytes of interest were well separated from other plasma constituents within 22 min at 45°C. The lowest detection limits of vitamins A and E and β-carotene were 0.02, 0.5, and 0.1 μg/mL, respectively. The recovery and reproducibility of the method were ∼90%. The method is sensitive, specific, and appropriate for epidemiological studies and routine determination of vitamin deficiency (Lee et al., 1992).
Isolated lymphocytes
Determination of H2O2 production
H2O2 generation in lymphocytes from climbers was assayed before and after the 21-day period in the high altitude environment using a colorimetric method involving the oxidation of iodide in the presence of ammonium molybdate, with photometric analysis of the resulting blue starch–iodine complex at 570 nm (M'Bemba-Meka et al., 2005). Briefly, human blood lymphocytes were treated with 38.5 mmol/L HCl, 80 mmol/L potassium iodide, 80 mmol/L ammonium molybdate in H2SO4, and 0.38% (w/v) starch. Twenty minutes after adding potassium iodide, sample absorbance was measured at 570 nm using a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA, USA). The H2O2 concentration was estimated using a standard curve. Results are expressed as milligrams H2O2 per 15 × 104 cells. For each experimental condition (pre- and postexpedition and follow-up) and for each sample derived from any subject, at least five wells were analyzed.
Determination of mitochondrial potential
Mitochondrial membrane potentials were determined using JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethylbenzimidazolylcarbocyanine iodide/chloride; Molecular Probes, Eugene, OR, USA), a cationic carbocyaninic dye that accumulates in mitochondria. When the transmembrane potential is high, as in normal cells, JC-1 forms dimers (J-aggregates) that emit red fluorescence; when the potential is low, an index of oxidative stress, the dye forms monomers that emit green fluorescence, and there is a concomitant decrease in red fluorescence. The red:green fluorescence ratio depends on the mitochondrial membrane potential and not on other factors, such as mitochondrial size, shape, or density, that might influence single-component fluorescence signals (Molecular Probes).
Isolated lymphocytes were loaded with 5 μg/mL JC-1 for 15 min at 37°C in growth media. At the end of the incubation, the cells were centrifuged, washed, and resuspended in normal external solution [NES: 140 mmol/L NaCl, 2.8 mmol/L KCl, 2 mmol/L CaCl2, 2 mmol/L MgCl2, 10 mmol/L glucose, 10 mmol/L 4-(2-hydroxyethyl)1-(piperazineethanesulfonic acid (HEPES); pH 7.3] and seeded at 5 × 103 cells/well in special-optics 96-well plates (Corning-Costar, Milan, Italy). Fluorescence was detected on a Gemini Spectramax XS fluorescence plate reader (Molecular Devices) using an excitation wavelength of 485 nm and recording the emissions of the JC-1 monomer and aggregate at 530 and 590 nm, respectively. For each experiment, the aggregate/monomer (red/green) ratios were plotted. For each experimental condition (pre- and postexpedition and follow-up) and for each sample derived from any subject, at least five wells were analyzed.
Glutathione peroxidase activity
Glutathione peroxidase activity was measured in samples derived from sonicated lymphocytes suspended in 100 mmol/L phosphate buffer (pH 7) containing 1.5 mmol/L dithiothreitol. Protein concentrations were determined using Protein Assay Kit (Bio-Rad Laboratories Srl, Milan, Italy) (Bradford, 1976), using bovine serum albumin (BSA) as a standard. Enzymatic activity was calculated according to the method of Paglia and Valentine (1967). The oxidation of nicotinamide adenine dinucleotide phosphate (NADPH) was monitored at 340 nm and 25°C. One unit of enzymatic activity was defined as the oxidation of 1 μmol NADPH/min.
Ca2+ Signaling Analysis
The intracellular Ca2+ content was monitored using the Ca2+-sensitive fluorescent indicator Fura-2-AM (Molecular Probes), and an inverted Olympus microscope connected to a high-speed wavelength switcher (Polychrome II, Till Photonics, Germany), equipped with a 75-W stabilized xenon lamp (Ushio Inc., Cypress, CA, USA) and a cooled, charge-coupled device (CCD) camera (C6790 model; Hamamatsu Photonics, Hamamatsu, Japan). Isolated lymphocytes (1 × 105 cells/mL) were loaded in suspension with 5 μmol/L Fura-2-AM (fura-2-acetoxymethyl ester) for 30 min at 37°C in NES, supplemented with 1% (w/v) BSA. Cells were centrifuged at 400 × g for 10 min and washed twice to remove extracellular dye. Next, cells were resuspended in fresh NES, transferred to special-optics 96-well plates (Corning-Costar) coated with poly-
Fura-2-AM-loaded lymphocytes were sequentially and repetitively excited at 340 and 380 nm; fluorescence images were acquired with a CCD camera and stored on an interfaced computer. The acquisition time was one image ratio per second. The image ratio calculations were carried out pixel by pixel on a pair of corresponding 340- and 380-nm image files. The temporal plots (mean value of the fluorescence signal in a selected cellular area) were calculated from the image ratios. [Ca2+]i in a single cellular field, recorded by a [Ca2+] calibration plot of the 340/380 ratio, was calculated using Calcium Calibration Kit for video imaging (Molecular Probes) (Pietrangelo et al., 2002). For each experimental condition (pre- and postexpedition and follow-up) and for each sample derived from any subject, at least five different wells were analyzed
Statistical Analysis
Statistical analyses, where indicated, were performed using Prism 4 for Windows (GraphPad Software Inc., San Diego, CA, USA). The data are presented as means ± SEM, as specified in the figure legends. Comparisons between groups were made using t-tests.
Results
The first results to emerge from the current study were obtained during isolation of lymphocytes from blood samples. Counts of isolated lymphocytes showed an increase in cell number in samples collected from climbers after the 21-day period of high altitude exposure relative to samples isolated before the expedition. Lymphocyte cell number returned to preexpedition values in samples obtained in the follow-up (Fig. 3).

Number of isolated lymphocytes in samples from the seven climbers before (Pre), soon after (Post), and 6 months after (Follow-up) the end of the expedition. The data are expressed as means ± SEM, n = 7. *p < 0.05 vs. Pre.
Three markers of oxidative stress, total antioxidant status, lipid peroxidation, and protein oxidation, were analyzed in sera isolated before and soon after the expedition, as described in Materials and Methods. These results are summarized in Fig. 4, which shows that none of these parameters was significantly different between pre- and postexpedition conditions, although lipid peroxidation levels measured after high altitude exposure were slightly higher than those measured at sea level.

Markers of oxidative stress in serum. Trolox-equivalent antioxidant capacity (TEAC), thiobarbituric acid-reactive substances (TBARS), and protein carbonyl content (PCC) were assayed in sera isolated from climbers before (Pre) and soon after (Post) the expedition. Data are expressed as means ± SEM, n = 7.
In the same serum samples, vitamins A, E, lycopene, and β-carotene were analyzed before and soon after the expedition. As shown in Fig. 5, values obtained after the expedition were not significantly different from those measured before the expedition, although the mean values found at the end of high altitude exposure did trend lower than preexpedition values.

Serum vitamin content. Vitamins A and E, lycopene, and β-carotene contents were assayed in sera isolated from climbers before (Pre) and soon after (Post) the expedition. Data are expressed as means ± SEM, n = 7.
The oxidative machinery was also assessed in lymphocyte cell populations by analyzing (1) live-cell H2O2 production and release, (2) live-cell mitochondrial potential, and (3) glutathione peroxidase activity in cell extracts. Figure 6A shows that release of H2O2 caused by cell oxidative activity was unchanged in samples collected soon after or 6 months after the end of the expedition compared with that in preexpedition samples. The enzymatic activity of glutathione peroxidase, a major antioxidant enzyme, was also unchanged (Fig. 6C). Interestingly, mitochondrial membrane potential was significantly reduced in lymphocytes isolated soon after and 6 months after the end of the expedition compared with basal (preexpedition) values (Fig. 6B).

Lymphocyte oxidative status. Lymphocytes isolated before (Pre), soon after (Post), and 6 months after (Follow-up) the end of the expedition were analyzed for (
[Ca2+]i was assayed in the same cell populations by video-imaging analysis of single cells. These assays revealed that mean basal [Ca2+]i was significantly higher in cells isolated soon after the expedition, but had returned to preexpedition values in cells collected at the 6-month follow-up (Fig. 7).

[Ca2+]i recorded in isolated lymphocytes. Basal [Ca2+]i in lymphocytes isolated before (Pre: n = 268 tested cells from 7 climbers), soon after (Post: n = 241 tested cells from 7 climbers), and 6 months after (Follow-up: n = 129 tested cells from 7 climbers) the expedition. The data are expressed as means ± SEM. ***p < 0.001 vs. Pre; ns, not significant.
The effect of the high altitude expedition on lymphocyte [Ca2+]i homeostasis was also confirmed by monitoring spontaneous intracellular Ca2+ oscillations in the same cell populations (Fig. 8A). In lymphocytes assayed before the expedition, spontaneous Ca2+ waves were recorded in about 26% ± 1.6% of cells; this percentage increased to 44% ± 2.0% and 38% ± 5.0% soon after and 6 months after the end of the expedition, respectively (Fig. 8B).
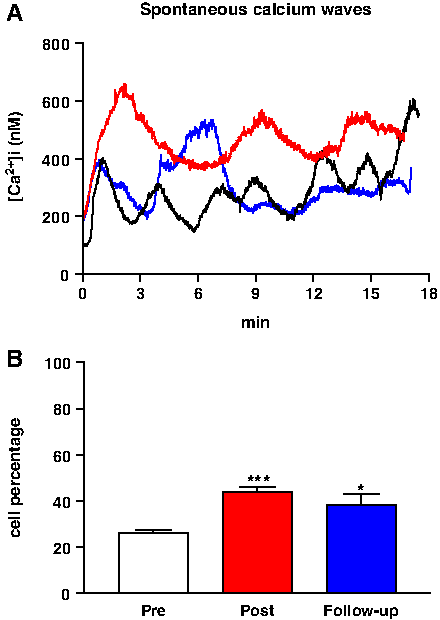
Spontaneous Ca2+ waves in isolated lymphocytes: (
Changes in [Ca2+]i in response to an oxidant stressor (H2O2) were also monitored by video imaging to assay the possible involvement of intracellular Ca2+ variation in the oxidative stress machinery. Addition of H2O2 (1 mmol/L) induced an increase in [Ca2+]i in a subpopulation of cells. In lymphocytes isolated before the expedition, about 30% ± 2.7% of cells were H2O2 responsive; this percentage trended slightly higher (36% ± 5.0%) in lymphocytes isolated soon after the expedition and significantly increased to 46% ± 6.0% in cells isolated at the 6-month follow-up (Fig. 9).

H2O2-induced changes in [Ca2+]i in isolated lymphocytes: (
Discussion
It is well known that a high altitude environment can affect various organ systems, notably the nervous, cardiovascular, and pulmonary systems, and the effects reflect modifications at the cellular level (Grocott et al., 2007; Hainsworth and Drinkhill, 2007; Hainsworth et al., 2007; Wilson et al., 2009). These effects result from the simultaneous influence of different environmental parameters, such as absolute altitude, humidity, temperature, climatic conditions, and duration of exposure. In addition, the level of physical activity needs to be taken into account. For these reasons, it is often difficult or unrealistic to attempt to directly correlate experimental results with a specific environmental or physical parameter. Accordingly, our goal was to provide a partial picture of the human response to such an unfavorable environment.
We used peripheral blood lymphocytes as biosensors of the whole-body response to high altitude and as a model for adaptation of the organism to environmentally induced modifications. Peripheral lymphocytes exhibit a number of important features. From an experimental point of view, primary lymphocytes can be easily and repeatedly collected. In addition, lymphocytes not only participate in inflammation mechanisms or as part of the immune response, but also constitute an important system that contributes to organ homeostasis and adaptation to pathological conditions and new environments (Walsh and Whitham, 2006; Zhang et al., 2007; Belia et al., 2009).
In addition, blood lymphocytes have also been used as biosensors of whole-body exposure to hypoxia, radiation, and physical activity, all challengers characterized by oxidative-stress-related phenomena (Niess et al., 1996; Moller et al., 2001; Lee et al., 2007). Consistent with previous reports (Klokker et al., 1993; Walsh and Whitham, 2006), we found an increase in lymphocyte number after high altitude exposure that returned to preexpedition values at the 6-month follow-up. Conversely, some authors found different responses in blood cell number and activation to hypobaric hypoxia, exercise, or both of these challengers and hypothesized that the observed effects depend on the cellular subpopulations and individual sensitivity to these stimuli (Chouker et al., 2005; Shtemberg et al., 2007; Wang and Lin, 2010). It is well known that lymphocytosis has resulted from different environmental conditions, such as variable physical activity levels, time exposure, and hypobaric hypoxia, probably owing to the mobilization of cells from the secondary lymphoid organs in response to stress (Hong et al., 2005).
It is possible to assess the functional status of lymphocytes under different conditions by evaluating cellular oxidative status and Ca2+ dynamics, which are important in regulating the functions of these cells and their ability to communicate with other blood cells (Li, 2008; Feissner et al., 2009). A reliable assessment of oxidative stress or damage in biological specimens requires a battery of assays that monitor oxidative status, associated changes in [Ca2+]i, and the levels of oxidatively modified macromolecules (Halliwell and Whiteman, 2004; Belia et al., 2009). As extensively reviewed by Hwang and Kim (2007), assays of malondialdehyde and protein carbonyl content are among the most widely used to determine lipid peroxidation and oxidative damage to proteins, respectively. Further, because ROS removal rate is controlled primarily by a variety of low-molecular-weight antioxidants, and considering that the net antioxidant effect reflects cooperation among a variety of enzymatic and nonenzymatic compounds with antioxidative properties, it is important to measure total antioxidant status in tissues and body fluids (Rice-Evans and Miller, 1994). Our data revealed that serum and lymphocyte markers that correlate with oxidative stress, tested here, were not significantly changed after hypobaric hypoxia exposure. These findings seem to contrast with other published results that show the activation of ROS generating systems and the weakening of enzymatic and nonenzymatic antioxidant systems, depending on the degree of altitude and time exposure (Dosek et al., 2007). The high altitude-induced oxidative unbalance can occur in different tissues. Indeed, this alteration may have relevance for the high altitude-induced microcirculatory dysfunction, as well as for pulmonary and cerebral edema (Gonzalez and Wood, 2001; Serrano et al., 2003). The macromolecular damage resulting from the hypobaric hypoxia-induced oxidative unbalance appears to be tissue specific. It has been reported that in rat skeletal muscles hypobaric hypoxia may induce lipid peroxidation and an increase of carbonyl derivatives, markers of oxidative protein damage, that conversely appeared reduced in the rat brain (Radak et al., 1994; Radak et al., 1997; Radak et al., 1998). In the Operation Everest III study, Joanny et al. (2001) reported that the levels of plasma lipid peroxidation were increased by 23% at simulated 6000 m and by 79% at simulated 8848 m, thus indicating that high altitude-induced oxidative stress is augmented as the severity of hypoxia increases. Another study showed that 3-month exposure to high altitude (4500 m) revealed increased lipid peroxidation and decreased enzymatic and nonenzymatic defense, whereas a 13-month sojourn induced normalization of redox balance (Vij et al., 2005).
In our study, we assayed samples before and soon after the expedition and did not observe any persistent unbalanced oxidative mechanisms or damaged molecules; on the other hand, we cannot exclude a possible transient increase in intracellular levels of ROS. Our data came from a real high altitude experience, and not from simulated hypobaric hypoxia. In addition, our study is based on a high-mountain expedition characterized by multiple parameters (such as gradual ascent and descent, moderate physical exercise, severe hypobaric hypoxia, and poor weather conditions) that could differently modulate the oxidative status of the climbers. The discordance between our results and those obtained by Joanny and co-workers may result also from the hormetic response triggered by high-impact and long-lasting aerobic exercise practiced by the subjects during the expedition. Indeed, it has been previously demonstrated that recurring bouts of nonexhausting physical efforts were able to enhance the main antioxidative protection systems and the activity of cellular machinery involved in the repair and detoxification molecular processes, as reviewed by Radak and colleagues (2008a).
The environmental conditions did, however, influence both [Ca2+]i and mitochondrial functions of lymphocytes. After high altitude exposure, the mitochondrial potential decreased; this effect appeared associated with an increase in [Ca2+]i, consistent with the strict linkage between levels of this ion and mitochondrial membrane permeability (Feissner et al., 2009). It is possible that this decrease in mitochondrial function reflects a compensatory response to oxidative stress. Alternatively, because the oxidative status of the cells was unchanged after high altitude exposure, this decrease might be independent of the oxidative stress machinery. Surprisingly, the lymphocyte mitochondrial membrane potential persisted at lower than preexpedition values during the follow-up period, a time when basal [Ca2+]i had returned to mean preexpedition values. Interestingly, the spontaneous Ca2+ wave activity of lymphocytes and their sensitivity to H2O2-induced increases in [Ca2+]i appeared to remain elevated at the 6-month follow-up point. During the 6-month period after the expedition, all subjects were still healthy, did not undergo any pharmacological therapy or treatment, and led a normal daily working life. For this reason, we cannot exclude that a posthypoxic reoxygenation process could be involved in inducing these results.
Because lymphocytes have a relatively long life-span, the cells are useful substrates for monitoring the incidence of cumulative, long-lasting stimuli (Balakrishnan and Rao, 1999; Gregoire et al., 2006). In addition, circulating lymphocytes experience tissue-specific and organ-function modifications induced by the environment. Over the lifetime of peripheral lymphocytes, transient responses of some parameters to expedition conditions recovered during the follow-up period. However, the cumulative impact of the environmental stimuli continued to be evident in certain residual modifications of lymphocyte function, here represented by an increase in spontaneous Ca2+ waves and enhanced cellular responses to the oxidative stressor H2O2.
Conclusions
The results obtained here reflect the aggregate responses of lymphocytes to the collective conditions that define the 21-day, high altitude expedition, including the effects of hypoxia, hypobaric pressure, temperature, humidity, and physical activity. In this study, high altitude conditions did not have a specific effect on global oxidative status as assayed by serum and lymphocyte markers. However, lymphocyte functions did show sustained effects of exposure, as revealed by changes in [Ca2+]i and Ca2+ signaling dynamics, as well as in mitochondrial membrane potential. Collectively, these findings suggest that mitochondria are the targets or source of the increased intracellular Ca2+ recorded in lymphocytes soon after the expedition. The importance of these observations lies in the fact that mitochondria represent the main metabolic system of the cell and serve a significant intracellular Ca2+ buffering function.
Footnotes
Acknowledgments
We wish to thank the seven climbers who enthusiastically participated in this study. In addition, the authors wish to thank Prof. G. Fanò, director of the Department of Basic and Applied Medical Sciences, for having sponsored the expedition and for helpful discussion of the results presented here. This work was also supported by research funds to Mariggiò and Amicarelli.
Disclosures
The authors have no conflicts of interest or financial ties to disclose.