
Abstract
Abstract
Brill, Anne-Kathrin, Alexander Kunz, Sebastian Robert Ott, Thomas Geiser, and Jacqueline Pichler Hefti. When “High” Is Not Very High: High Altitude Pulmonary Edema as First Manifestation of Sarcoidosis-Related Pulmonary Hypertension. High Alt Med Biol 13:285–287, 2012.—High altitude pulmonary edema (HAPE) is a life-threatening complication of high altitude stay, which may occur above altitudes of 2500 m. This is a case report of a healthy 41-year-old man, presenting with recurrent HAPE at moderate altitude. Medical work-up revealed an idiopathic pulmonary artery hypertension (PAH), and specific vasoactive treatment was started. Despite treatment with an endothelin receptor antagonist, the patient deteriorated clinically. Subsequent medical reevaluation showed a significant progress of mediastinal lymphadenopathy. Due to the histological proof of sarcoidosis, the initial diagnosis of PAH had to be changed to sarcoidosis-related pulmonary hypertension. Initiation of immunosuppressive therapy with corticosteroids led to significant and clinically relevant decrease in pulmonary artery pressure, even allowing episodes of asymptomatic re-exposure to moderate altitude. This case describes HAPE as first manifestation of a sarcoidosis–related pulmonary hypertension with a very unusual and early presentation of the underlying disease in an apparently healthy mountaineer.
Introduction
The Case
We present the case of a 41-year-old male Swiss, who reported recurrent dyspnea during mountaineering and hunting trips in the last 15 years. He had a medical history of four episodes of HAPE at altitudes of approximately 2200 m, 2800 m, 3400 m, and 4000 m, respectively. Two episodes of HAPE were confirmed by chest x-ray. The other episodes were diagnosed clinically as the patient developed the identical typical symptoms of HAPE after ascent and did not seek medical advice immediately. After his fourth episode of HAPE, he received a diagnostic work-up in 2007, including right heart catheterization at an altitude of 550 m, which revealed pulmonary arterial hypertension with an elevated mPAP of 34 mmHg (sPAP 50 mmHg, wedge pressure 15 mmHg, cardiac output 6.0 l/min, pulmonary vascular resistance 253 dyn*sec/cm5) not responding to intravenous iloprost and with an exercise-induced elevation of mPAP up to 57 mmHg. Clinical (oximetry breathing room air at rest 98%, BP 125/80 mmHg, heart rate 68 bpm) and laboratory parameters (hemoglobin 158 g/l, TSH, HIV, liver enzymes), as well as V/P scans, were normal. Lung volumes and CO diffusion capacity were within the normal range. Six-minutes walk test showed a walking distance of 620 m with a desaturation from SaO2 97% to 90%. Extensive search for underlying diseases as cause of pulmonary hypertension (PH) revealed a mild mediastinal lymphadenopathy on chest CT scan, only. Transbronchial needle aspiration (TBNA) of the mediastinal lymph nodes did not show any specific pathology. Reactive lymphadenopathy was assumed and PAH diagnosed. One year after diagnosis, clinical symptoms worsened with progressive dyspnea on exertion, and the repeated right heart catheter examination showed an increase in mPAP to 47 mmHg (sPAP 66 mmHg, wedge pressure 12 mmHg, cardiac output 5.7 l/min, pulmonary vascular resistance 491 dyn*sec/cm5) with an exercise-induced elevation of mPAP up to 80 mmHg. Specific treatment with the endothelin receptor antagonist bosentan was initiated with a satisfactory clinical and echocardiographic response (improvement of performance status, reduction of echocardiographically measured sPAP to 55 mmHg).
Two years later, the patient clinically deteriorated (functional class III) despite treatment with bosentan (2× 125 mg). PAP had increased again to sPAP of 70 mmHg measured by echocardiography. He was transferred to our clinic for reevaluation of treatment of pulmonary hypertension. Diagnostic reassessment showed a massive progression of the mediastinal lymphadenopathy (Fig. 1) and endobronchial ultrasound guided TBNA revealed noncaseating epitheloid cell granulomas confirming the diagnosis of sarcoidosis.
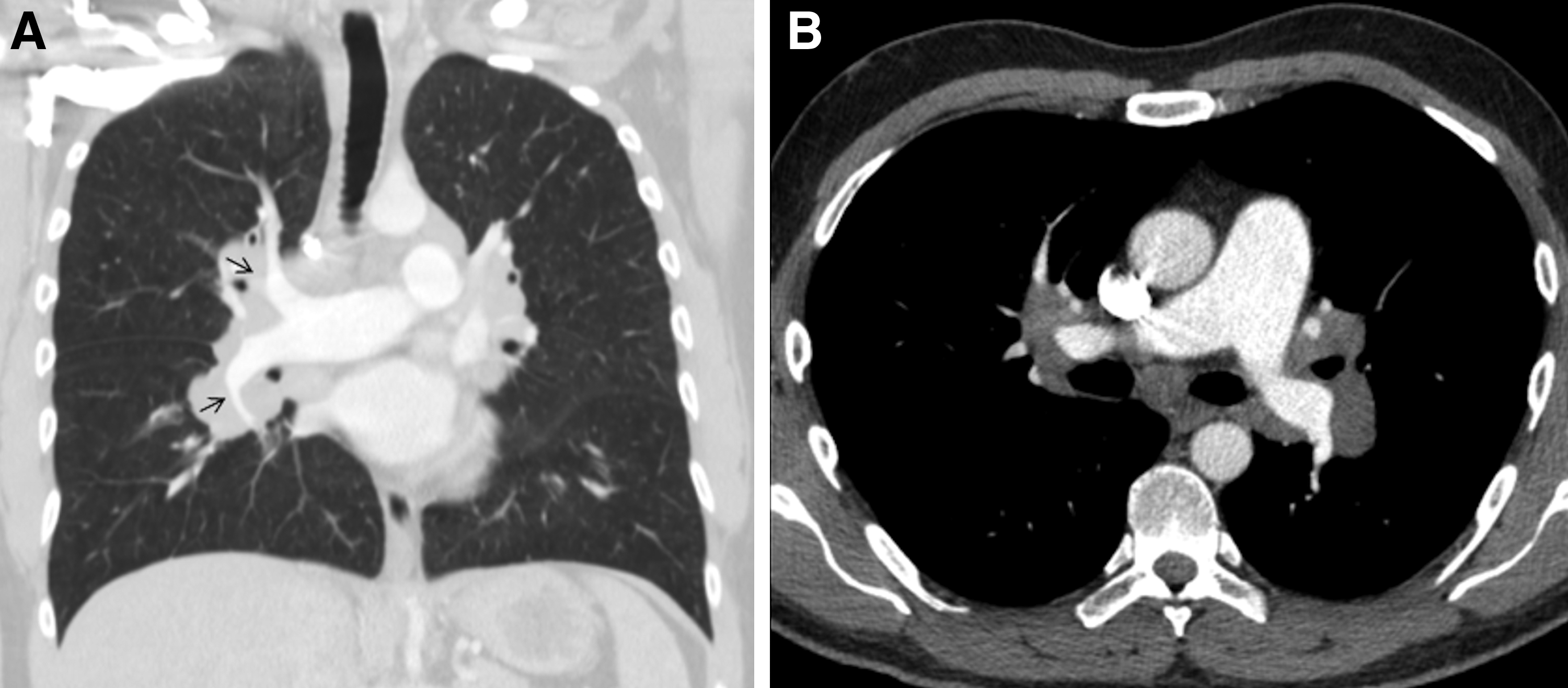
Mediastinal lymphadenopathy surrounding the central pulmonary arteries at point of diagnosis of sarcoidosis
An immunosuppressive treatment with systemic corticosteroids (starting dose 0.5 mg prednisone/kg body weight, followed by slow dose reduction) was added to bosentan. Subsequently, sPAP (echocardiographically assessed 45 mmHg) and extent of lymphadenopathy, compressing the pulmonary arteries, significantly decreased after 3 months. Clinical symptoms improved to functional class II.
Unfortunately, the patient stopped his medication without medical consultation due to personal reasons after 12 months of successful treatment with a suspected subsequent increase in PAP. The passionate mountaineer re-exposed himself to an altitude of 1800 m, which caused another severe episode of pulmonary edema. Rapid descent did not reverse symptoms and admission to an ICU was needed because of severe hypoxemia and progressive right heart failure (sPAP 90 mmHg, pulmonary artery catheter). Despite intensive therapy, right heart failure proceeded and the patient died.
Discussion
Sarcoidosis-related pulmonary hypertension (SAPH) is overall a rare form of pulmonary hypertension (Gluskowski et al., 1984). As SAPH is associated with a substantial increase in mortality (5-year survival of 59%), early diagnosis and more aggressive treatment are crucial (Nunes et al., 2006). SAPH can occur either by the formation of vascular granulomas or by mechanical compression of the pulmonary artery vessels by mediastinal lymphadenopathy. Usually, TBNA is a sensitive method to diagnose sarcoidosis in mediastinal lymph nodes (Agarwal et al., 2012). In our patient, the unspecific first biopsies may represent a sampling error and misled to the diagnosis of PAH.
Diagnosis of PAH should be carefully reevaluated if individual therapeutic goals are not achieved, as treatment options of pulmonary hypertension might alter according to possible underlying disease. In this case, HAPE was the early manifestation of PH in an apparently healthy subject, and retrospectively the initial treatment with bosentan was an inappropriate choice.
HAPE is reported in less than 0.01% of mountaineers. Several studies and clinical experience indicate the presence of HAPE-susceptibles, that is, subjects who show an enhanced hypoxic pulmonary vasoconstriction and who are therefore at higher risk for developing HAPE, even at lower altitude (Kawashima et al., 1989). This case, where incipient pulmonary hypertension was the reason for HAPE susceptibility, highlights the possible impact of preexisting disease in HAPE-susceptibles, which has been demonstrated previously by other case reports (Naeije et al., 1996; Schoene, 2001; Roggla et al. 2007;). It is still not clear, when “high” is high enough to serve as satisfying trigger for a pulmonary edema to be an “idiopathic” HAPE. Therefore, we believe that a closer work-up of individuals developing HAPE at moderate altitude is needed to exclude underlying pathology. Long-term cohort studies are necessary to evaluate whether HAPE-susceptibles are at increased risk for developing pulmonary hypertension due to any reason.
Footnotes
Author Disclosure Statement
AKB, AK and JPH do not have any conflicts of interest to disclose. SRO received money for teaching activities and talks from Bayer and Pfizer. TG received money for teaching activities and talks from Novartis and Actelion.