
Abstract
Abstract
Martha C. Tissot van Patot, Martha C., German Ebensperger, Max Gassmann, and Aníbal J. Llanos. The hypoxic placenta. High Alt Med Biol. 13:176–184, 2012. —Hypoxia of the placenta is integral to complications of pregnancy, including preeclampsia, intrauterine growth restriction, and small-for-gestational age babies. Hypoxia in the placenta is associated with vascular remodeling, hypertension, metabolic changes, oxidative stress, mitochondrial dysfunction, and endoplasmic reticular stress. Hypoxia induces similar outcomes in other organs such as the lungs, kidney, and gut. Comparing and contrasting the effects of hypoxia on placental functions and functions of lung, kidney, and gut can lead to novel hypotheses and investigations, furthering our understanding of the impact of hypoxia on these diverse yet similar organs. In this review, we compare and contrast hypoxic placental responses to those in the other organ and cell systems.
Introduction
Hypoxic Responses as in the Lung
Vascular remodeling
Hypoxia causes pulmonary and uteroplacental arteries to have greater vasoconstrictive capabilities than during normoxia, and both vessels are associated with the development of hypertension. Note that the term ‘vascular remodeling’ has led to confusion, as it has different meanings depending as whether this term is used in pulmonary or placental areas of research. In pulmonary research, ‘vascular remodeling’ describes the pathophysiologic vascular changes induced by hypoxia, while in placental research it is used to describe physiologic changes during normoxic exposure. The impact of hypoxia on placental and pulmonary vasculature, however, is very similar.
There are two distinct circulations in the placenta, maternal and fetal. The maternal circulatory system undergoes remodeling or ‘physiological conversion’ during development. In a healthy pregnancy, as the placenta implants into the uteroplacental wall and oxygen tension increases with proximity to the uteroplacental arteries, the involved arteries are ‘remodeled’. This means that originally muscularized and vasoreactive vessels become now dilated, nonmuscularized, and nonvasoreactive. This process insures sufficient blood flow to the placenta and in turn efficient oxygen and nutrient delivery to the fetus (Fig. 1).
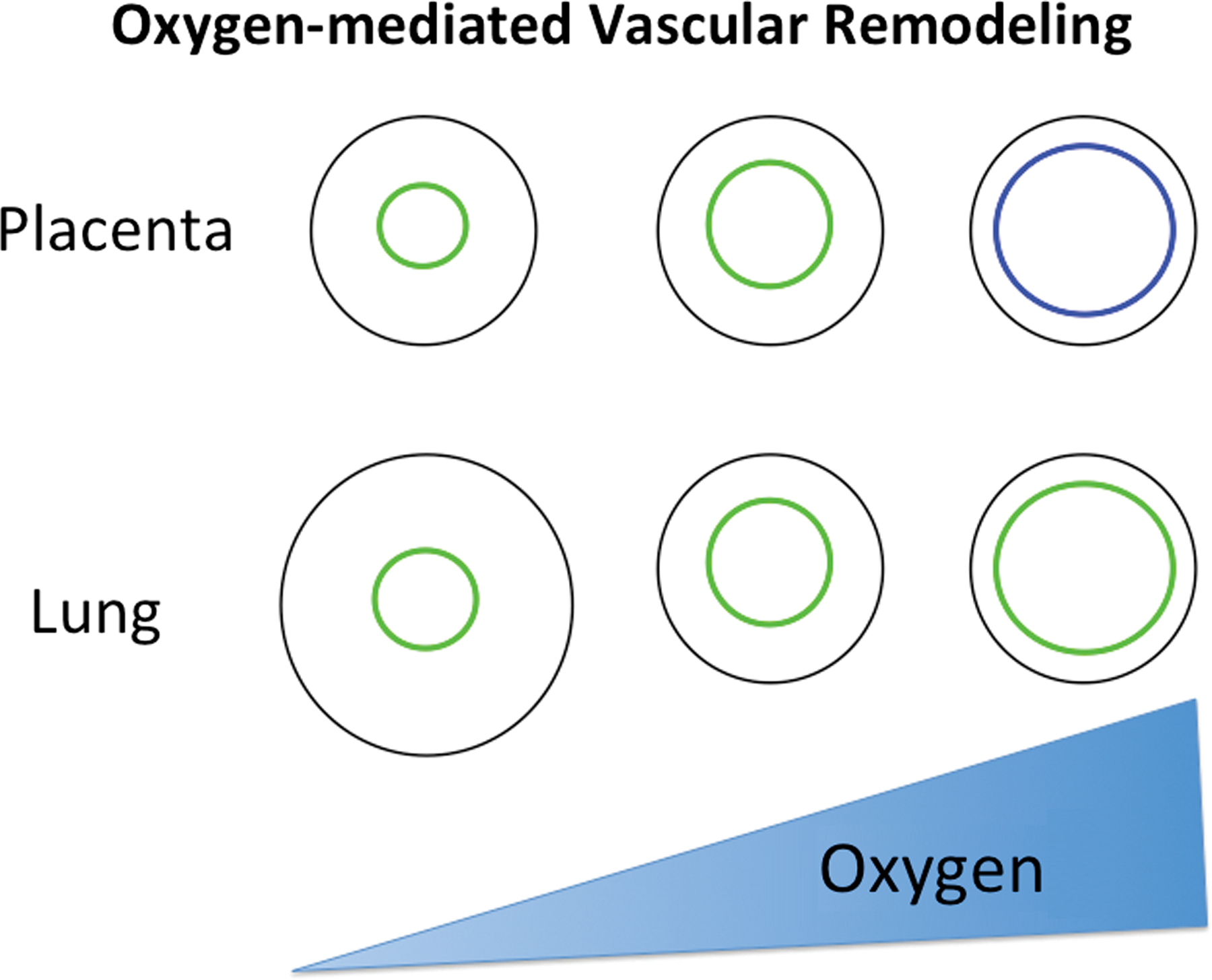
The relationship of oxygen to vascular phenotype in placenta and lung is depicted. The relative values for low and high oxygen are related to the ‘normoxic’ oxygen for each organ, relatively low pO2 in the placenta and relatively high PO2 in the lung. Low oxygen in these organs yields vasoconstricted vessels, and the pulmonary system has a remodeled medial layer to be thickened primarily with smooth muscle cells. Higher oxygen concentrations yield dilated vessels in the placenta and lung, and at the maternoplacental interface endothelial cells are replaced by nonvasoreactive trophoblast. Green depicts endothelial cells, blue depicts trophoblasts.
When a placenta is exposed to lower pO2 during invasion, as occurs in residents at high altitude or in anemic patients, physiologic placental remodeling is impaired, resulting in the presence of more muscularized and vasoreactive vessels (Tissot van Patot et al., 2003; Cartwright et al., 2007). The process of physiologic remodeling in the placenta involves release of a variety of cytokines, matrix metalloproteinases, and growth factors by the placenta's decidual cells, thereby causing dilation of the vessels, followed by invasion of trophoblasts, specialized placental epithelial cells, and uterine natural killer cells (Harris, 2011). In a fully remodeled uteroplacental artery, nonvasoactive trophoblasts replace endothelial cells, and contractile smooth muscle cells are no longer present. In term placenta, endothelial cells can be found in some uteroplacental vessels of the basal plate, not only lining the artery but extending onto the inner surface of the basal plate (Ockleford, 2010). It is unclear if the uteroplacental arteries are re-endothelialized near term, thereby providing a means to reduce blood flow during placental delivery or if the vessels were never fully remodeled.
To come back to our analogy to the lung: many of the factors governing trophoblast invasion and uteroplacental remodeling are also involved in hypoxia-induced pulmonary vascular remodeling including, but not limited to, HIF alphas, VEGF, PlGF, IL-6, TGFβ, and MAP kinases (Stenmark et al., 2006; Hanze et al., 2007; Knofler et al., 2011) (Table 1). Pulmonary vascular remodeling begins with hypoxia-induced vasoconstriction, followed by medial and adventitial thickening. Increased vessel wall thickness can be attributed to the presence of an elevated number of smooth muscle cells, many of which are hypertrophied, and excessive collagen and elastin deposition (Stenmark et al., 2006) (Fig. 1).
References are primarily reviews discussing the role of the specific factor in the placenta or lung during hypoxia.
Hypertension
Hypoxia is well known to result in hypertension in the pulmonary system and in the placental and maternal circulations. Notably, hypertension is prevalent in pregnancies at high altitude (Moore et al., 1982a; Palmer et al., 1994), leading a high altitude clinician to comment that “a pregnancy without high blood pressure is notable” (Lisa Zwerdlinger, M.D., Leadville, Colorado). Indeed, the placenta plays a key role in the development of maternal hypertension by releasing multiple vasoconstrictive factors, among those, reactive oxygen species (ROS), HIF-1α, lowered nitric oxide, increased sensitivity to angiotensin II, endothelin-1, and arachadonic acid metabolites such as thromboxane (Gilbert et al., 2008), all of which have also implicated in hypoxia-induced pulmonary hypertension (Sommer et al., 2008; Ward et al., 2009) (Table 1).
Hypoxic Placenta Outcomes on Fetal/Neonatal Lung
Because the placenta is ultimately responsible for the nurturing of the fetus, it is interesting in our analogy to consider what happens to the fetal pulmonary vascular system in response to placental hypoxia. Recently, Jayet et al. (2010) reported that children from preeclamptic pregnancies had 30% greater pulmonary vascular pressure than children from normotensive pregnancies. In support of these findings, it was found that the offspring of sheep exposed to high altitude during pregnancy have more muscularized pulmonary arteries and greater pulmonary vascular pressure than offspring from low altitude pregnancies (Llanos et al., 2011). Furthermore, pulmonary arteries demonstrated greater sensitivity to vasoconstrictors, KCl, and endothelin-1, similar to the augmented responses of hypoxic placental and pulmonary vessels.
Taken together, the molecular and biochemical mechanisms involved in the hypoxic responses of placental and pulmonary vessels have extensive overlap. Detailed comparisons between the hypoxic responses of these organ systems deserve close scrutiny in lieu of the fact that the common final outcome, muscularized, hyperreactive, vasoconstrictive vessels, is associated with diseases that have high incidences of morbidity and mortality, such as preeclampsia and pulmonary hypertension. Indeed, the roles of RhoA and RhoA kinase (ROCK) in the hypoxic lung, but not placenta, have been extensively researched in recent years. It is now clear that the RhoA pathway plays a critical role in the pulmonary response to hypoxia (Ward et al., 2009). The few investigations in placenta demonstrate that RhoA/ROCK may be involved in trophoblast migration (Fafet et al., 2008; Nicola et al., 2008), but further investigations are necessary to clearly delineate the role of RhoA/ROCK in the hypoxic placenta.
Conversely, the TRAIL–FASL-mediated apoptotic pathway has been implicated in compromised placental vascular remodeling during hypoxia (Whitley et al., 2010), but not in hypoxia-mediated pulmonary vascular remodeling. Interestingly, TRAIL has been implicated in pulmonary fibroblast expression of TGFβ and collagen production (Yurovsky, 2003). These examples provide support for the value of comparing and contrasting placental responses to hypoxia with responses of the pulmonary system.
Hypoxic Responses as in the Kidney
The hypoxic placenta produces factors usually considered the primary domain of the hypoxic kidney, including erythropoietin (Epo), angiotensin II, and adrenomedullin (ADM) (Conrad et al., 1996; Marinoni et al., 2007; LaMarca et al., 2011). For decades it was understood that hypoxia induces renal Epo production, thus stimulating erythropoiesis. In recent years, it has become clear that Epo has many functions other than stimulating the production of red blood cells. Nonerythroid functions include inhibition of apoptosis and stimulation of inflammatory and pro-angiogenic mechanisms in multiple organ systems, including the central nervous system, cardiovascular system, auditory system, retinal system, and the pancreas (Grimm et al., 2002; Gassmann et al., 2003; Chateauvieux et al., 2011). These nonerythroid actions of Epo serve to protect tissues from hypoxia and ischemic/reperfusion injury (Mihov et al., 2009; Pappo et al., 2010).
In response to hypoxia, the placenta is also highly capable of Epo production, and has been reported to synthesize up to 20 times more Epo than the fetal kidney (0.44 vs. 0.18 million mU per hour) in an ovine model (Davis et al., 2003). Other than increasing fetal hemoglobin, nonhematopoietic Epo actions have the potential to positively or adversely affect placental and fetal development. For example, apoptosis is a key physiologic mechanism in normal fetal development and uteroplacental artery invasion and remodeling. Epo's pro-angiogenic actions might improve placental vascularity, thus elevating gas exchange between fetus and mother during hypoxia. Further, ischemic reperfusion injury (Burton et al., 2004) and elevated placental Epo concentrations (Teramo et al., 2004) have been associated with preeclampsia, a disease of pregnancy that involves reduced oxygen supply to this organ. Reports of Epo-mediated protection of ischemic reperfusion injury in heart and liver (Mihov et al., 2009; Pappo et al., 2010) suggest that elevated placental Epo production may be of benefit in preeclampsia.
Adaptation of highlanders
In the kidney, hypoxia-induced Epo production is primarily mediated by hypoxia inducible factor −2 (HIF-2) (Rankin et al., 2007). In the placenta, hypoxia led to greater accumulation of HIF-2α but not of HIF-1α (Genbacev et al., 2001, Tissot van Patot et al., 2004). Interestingly, polymorphisms of EPAS1, the gene encoding HIF-2, were recently reported in Tibetans highly adapted to residence at extreme altitudes. These polymorphisms were highly associated with low hemoglobin concentration in the Tibetan population (reviewed in Tissot van Patot et al., 2011). In other words: the kidney likely has less HIF-2/Epo expression in this adapted population. Tibetans also experience a much lower incidence of preeclampsia and low birth weight at high altitude, compared with the Han population, who represent Chinese native to low altitude (Julian et al., 2009). Placental function is intimately involved in the development of preeclampsia and birth weight, and Epo is implicated in the development of preeclampsia and reduction of body weight (Katz et al., 2010). It is quite possible that the EPAS1 adaptation extends to placentas from healthy, but not preeclamptic or low birth weight, pregnancies at high altitude.
EGLN1 is the gene encoding prolyl hydroxylase 2 (PHD2) that in turn inhibits the α subunits of HIFs by targeting them for ubiquination. PHD2 is specific for inhibition of Epo production in kidney (Minamishima et al., 2010). Severe placental defects in PHD2-deficient mice result in highly elevated HIF concentrations and embryonic lethality between E12.5–E14.5 (Takeda et al., 2006). Interestingly, a mutation of the EGLN1 gene is reported in Andean populations (Peng et al., 2011) who are well adapted to high altitude and—unlike the Tibetans—Andeans such as Quechuas and Aymaras have high hemoglobin concentrations (Beall et al., 1998). These data suggest that the EGLN1 mutation lowers PHD2, leading to elevated renal production of HIF2 and Epo. Accordingly, Andeans do not demonstrate the mutation in the EPAS1 gene that is present in Tibetans (Beall et al., 2010).
In summary, genomic analyses suggest that Tibetans have adapted to altitude by mutating EPAS1, reducing HIF-2, Epo, and hemoglobin concentrations, while Andeans have adapted by mutating PHD2, increasing HIF-2, Epo, and hemoglobin concentrations. Such variability in placental Epo concentrations would result in greatly differing placental morphology and functions between these adapted populations. Further studies are urgently needed to discover how such differing genetic mutations can each result in successful adaptations and pregnancies in high altitude native populations.
Impact on blood pressure
Hypoxia not only increases renal and placental Epo production, but hypoxia or Epo alone induces activity of the renin–angiotensin system (RAS) in a variety of ways. The RAS increases blood pressure directly via receptors in the vascular system and indirectly by increasing blood volume via retention of sodium in the kidney. In the placenta, Epo increases vasoconstriction via the angiotensin I (AT1) receptor primarily in placental veins delivering blood to the fetus (Resch et al., 2003). Placental hypoxia increases angiotensin II production (Knock et al., 1994), activity of angiotensin converting enzyme (ACE) (Ito et al., 2002), and AT1 receptor agonist autoantibodies (Wallukat et al., 1999), all of which are elevated in preeclamptic placentas (Herse et al., 2008). Activation of angiotensin receptors in the placenta increase vasoconstriction of veins delivering blood to the fetus (Resch et al., 2003), decreases trophoblast differentiation and invasion (Araki-Taguchi et al., 2008), increases reactive oxygen species (ROS) via NADPH oxidase and increases NFκB and thus inflammation (Dechend et al., 2003), all of which are implicated in preeclampsia.
In the kidney, hypoxia and/or Epo stimulate an increase in angiotensin II, which in turn leads to an increase in systemic blood pressure. As in the hypoxic placenta, angiotensin II can also cause cellular damage by initiating oxidative stress (Palm et al., 2011). Thus, the RAS has been implicated in the development of renal allograft rejection, renovascular disease, and malignant hypertension. Interestingly, AT1 receptor agonist autoantibodies are elevated in these conditions and blocking receptor binding can reduce blood pressure and kidney damage (LaMarca et al., 2011). Angiotensin II blockade or ACE inhibition can also reduce blood pressure and kidney damage (Lebel et al., 2006), however the hematocrit also falls in patients treated this way (Qureshi et al., 2007). Delivery of ACE inhibitors during pregnancy is not advised due to increased risk of major congenital malformations (first trimester) and elevated risk of fetopathies (second and third trimesters) (Cooper et al., 2006).
As expected by the highly variable data from hypoxic kidneys cited above, epidemiologic studies of kidney function at high altitude have variable results. Some studies show altitude residents having a greater disposition toward a syndrome referred to as high altitude renal syndrome (Arestegui et al., 2011) and development of chronic kidney disease (Chen et al., 2011), while others demonstrate a beneficial effect of altitude by decreased mortality in dialysis patients (Winkelmayer et al., 2009) and a protective effect in chronic kidney disease patients (Ghahramani et al., 2011). The kidney and placenta play important roles in control of blood pressure in response to hypoxia. However, the hypoxic responses also cause damage to the cells and organ systems. The similarities between these two organ systems in their mechanisms of hypoxic response go far beyond those that are highlighted here and include nitric oxide, generation of reactive oxygen species, and changes in mitochondrial function. The resulting hypertension and end organ dysfunction have high rates of mortality and there is great potential for similar therapeutic interventions to work on both organ systems.
Hypoxic Responses as in the Gut
Placental function mimics gut function by selectively delivering nutrients from the maternal circulation to the fetus. Hypobaric hypoxia results in lower birth weight (Lichty et al., 1957; Moore et al., 1982b) and loss of weight in response in adults (Westerterp et al.,1992; Lippl et al., 2010). Unfortunately, present data regarding placental expression of glucose transporter 1 (GLUT1) during hypoxia are variable and inconclusive (Hayashi et al., 2004; Zamudio et al., 2010). It is likely that the reduction in fetal glucose concentrations at high altitude, associated with lower birth weight, is due to a combination of factors. Moore et al. (2011) have demonstrated significant altitude-associated reduction in uterine artery blood flow, thus delivering less glucose to the placenta and fetus, while Zamudio et al. (2010) suggest that the placenta increases glucose uptake, thus ‘robbing’ the fetus of glucose at high altitude. Recent metabolomics data, however, do not reveal direct evidence of greater placental glycolysis in high as compared to low altitude placentas, yet altitude placentas did demonstrate a tendency toward storing energy in the form of phosphocreatine (Tissot van Patot et al., 2010). Furthermore, placental hormonal factors, including leptin and ghrelin, are associated with low birth weight in preeclampsia (Aydin et al., 2008) at high altitude (Trollmann et al., 2007).
Similar to the reduction in birth weight when pregnancy occurs at high altitude, ascent to altitude in human subjects results in reduced body weight. The mechanism of weight loss at altitude remains unknown, yet gastrointestinal blood flow does not appear to be a factor (Kalson et al., 2010), while the roles of leptin and ghrelin are disputed (Quintero et al., 2010; Aeberli et al., 2012). These authors discuss additional mechanisms of hypoxia-induced weight loss that have been implicated, including loss of appetite, poor nutrient absorption, and inflammation (Quintero et al., 2010), some of which deserve investigation in the phenomenon of low birth weight at high altitude.
The Common Denominator: Oxidative Stress
Hypoxia results in oxidative stress, generation of reactive oxygen species (ROS), and initiation of multiple molecular cascades, resulting in a myriad of biological outcomes, including hypertension, vascular remodeling, erythropoiesis, angiogenesis, glycolysis, and many others. ROS can activate physiologic processes to protect cells from hypoxia as well as pathologic process that damage organs. These molecular cascades initiated by ROS include activation of a variety of transcription factors and protein kinases, opening ion channels, protein modifications, lipid peroxidation, and DNA oxidation (Burton et al., 2011). In pathological conditions, ROS-initiated pathways can lead to vascular remodeling, hypertension, altered cellular differentiation or proliferation, impaired cellular transport, reduced enzyme kinetics, and many more. Chronic exposure to ROS impacts the placenta and is involved in the development of intrauterine growth restriction, preeclampsia, and spontaneous miscarriage (Burton et al., 2011), largely by impairing uteroplacental artery remodeling, uterine invasion, and vascular development. Similarly, the effects of ROS on pulmonary, renal, and gastrointestinal systems can contribute to the development of pulmonary hypertension, chronic kidney disease, and inflammatory bowel disease.
Mitochondria generate ROS during hypoxia, which in turn mediate cellular response to hypoxia (Hamanaka et al., 2010). The complexity of the system and overlap in function is extreme. For example, mitochondrial ROS activate stabilization of hypoxia-inducible transcription factors (HIFs) (Fandrey et al., 2006) and HIFs mediate mitochondrial respiration during hypoxia (Chandel et al., 2000). There is also a close relationship between oxidative stress, mitochondrial response, and the endoplasmic reticulum (ER). Because protein metabolism is responsible for about 30% of cellular oxygen consumption, it is vital for the ER to communicate with the mitochondria during hypoxic stress (Burton et al., 2011). In the placenta, it was recently reported that hypobaric hypoxia created ER stress, activating the unfolded protein response, thus modulating protein synthesis in placentas, possibly contributing to the reduced villous volume in high altitude placentas and the resultant lower birth weight (Yung et al., 2012). Hypoxic activation of the unfolded protein response in endoplasmic reticulum has also been reported in hypoxic kidneys (Inagi, 2010) and endothelium (Bouvier et al., 2009).
Translational science: How does the hypoxic placenta affect the fetus?
When hypoxia creates pathologies in the lungs, kidney, and gut, the patient suffers illness and disease. When hypoxia creates pathophysiology in the placenta, the fetus may suffer developmental pathologies. It has recently been reported that children from preeclamptic pregnancies at high altitude have elevated pulmonary artery pressures (Jayet et al., 2010). Similarly, we have reported elevated pulmonary artery pressures and differences in responses to vasoactive substances in an ovine offspring from high altitude pregnancies (Herrera et al., 2010; Llanos et al., 2011; Moraga et al., 2011). These data strongly suggest that a hypoxic placenta produces long-lasting changes in fetal physiology.
We have recently extended these studies to the cardiovascular system, hypothesizing that the cardiovascular basal status and cardiovascular defense responses to acute hypoxia in lowland fetal sheep grown at high altitude are blunted in comparison to fetuses that underwent gestation at sea level. Using a protocol similar to that previously reported (Herrera et al., 2010; Moraga et al., 2011), we studied fetal at day 130 of gestation (87% gestation) at 580 meters above sea level (LALA) and fetal sheep from lowland taken to 3600 meters above sea level at 45–50 days of gestation (30% gestation) (LAHA). To summarize our findings, in fetuses whose gestation took place partially in chronic hypoxia (LAHA) we found that:
1. Three out of 7 LAHA fetuses died in utero prior to the age of catheterization and there was marked fetal growth retardation. In contrast, all the sea level pregnancies (LALA) had 100% survival. The striking decrease in PO2 in LAHA fetal sheep can explain the marked fetal growth restriction and mortality in these fetuses (Table 2). 2. No bradycardia was seen as response to hypoxia. In contrast, there was an increased heart rate, evidencing an absent chemoreflex with a diminished vagal response plus an augmented sympathetic outcome (Fig. 2). 3. Basally, there was an increased carotid blood flow, as the result of the low PO2 in the arteries supplying the central nervous system, probably due to an increased NO production, among other mediators (Fig. 3). 4. The systemic arterial pressure and carotid blood flow showed blunted responses to acute hypoxemia, responses observed even during the recovery period (Figs. 2 and 3).
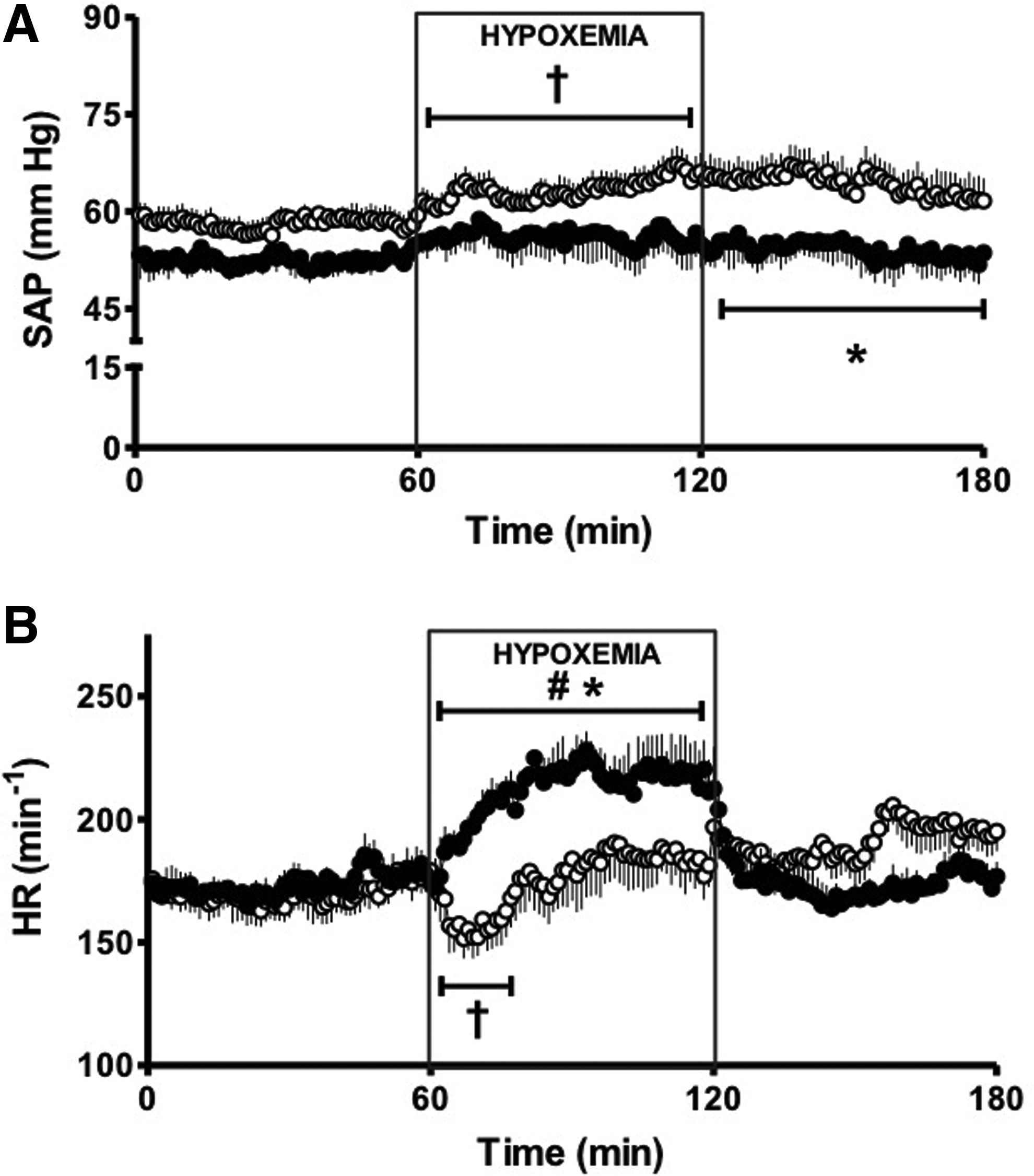
Mean systemic arterial pressure (SAP;
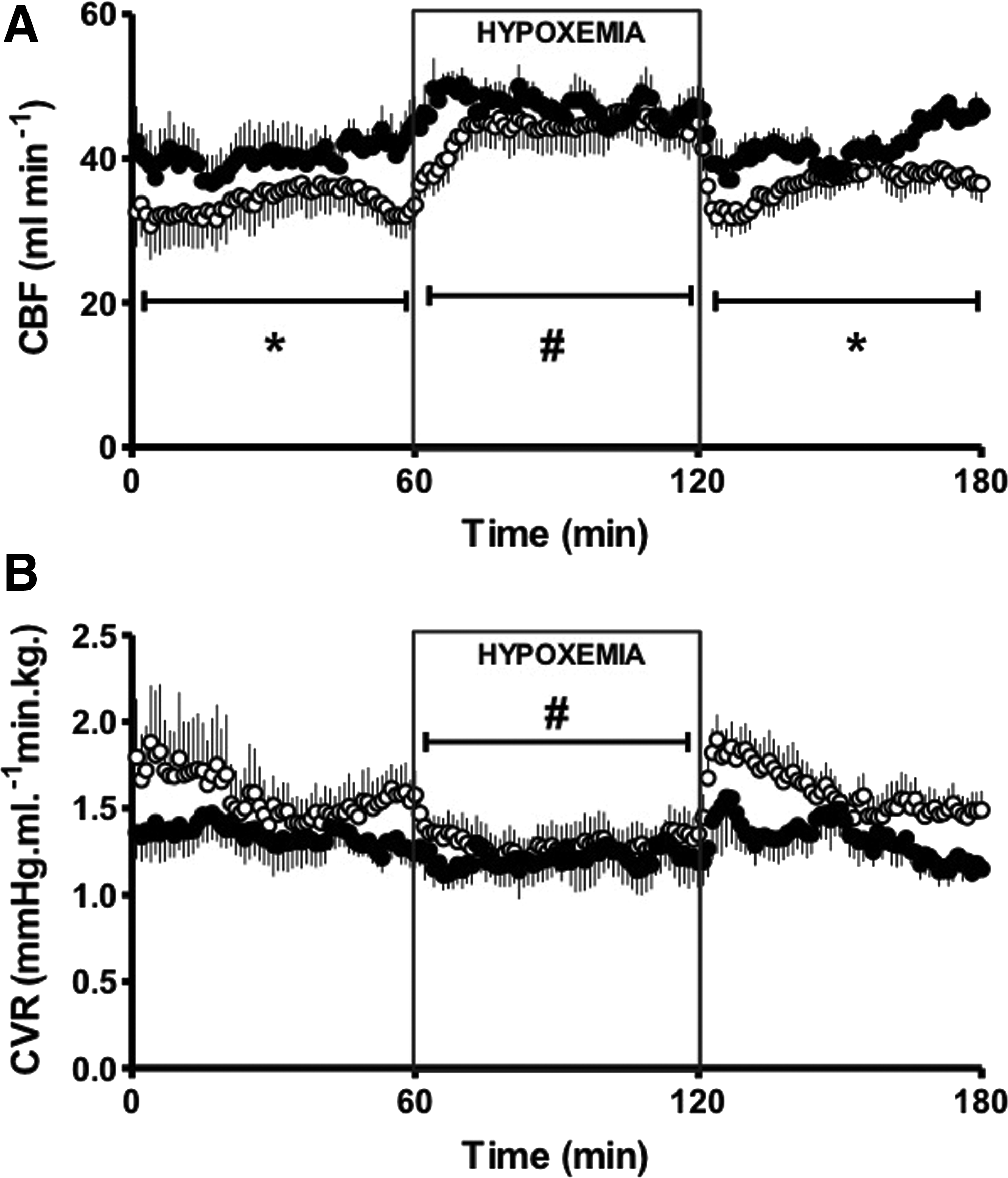
Carotid blood flow (CBF;
Values are means±SEM taken at 15 and 45 min of basal, each 15 min during hypoxemia, and 15 and 45 min of recovery. Significant differences (p≤0.05).
†, vs. basal period; #, vs. all periods in the same experimental group; *, vs. LALA fetuses.
We speculate that the observed blunting in the cardiovascular responses in chronically hypoxic fetuses reflects the expression of a hypometabolic state in the tissues, fingering the main regulation at cellular and tissue levels with little need of major cardiovascular adjustments to cope with acute changes in PO2.
These data support the hypothesis that a hypoxic placenta can have a strong influence on the health of the fetus, similar to the way in which hypoxic organs negatively impact the health of the individual.
Conclusion
In its role of supporting fetal development, the placenta must fulfill the actions of many physiologic processes, including the delivery of oxygen and nutrients, removal of waste products, blood pressure control, and hormone production. Hypoxia stresses the placenta's ability to perform these functions in a manner similar to hypoxia-induced stress created in other organs responsible for similar functions such as the lungs, gut, and kidneys. Furthermore, at the cellular level, hypoxia-induced oxidative stress activates similar cascades in trophoblast and vascular endothelial cells, smooth muscle cells, and fibroblasts in multiple organ types. Comparing and contrasting these molecular cascades and the resulting hypoxia-induced pathophysiologies in various organ systems will likely stimulate further investigations into the etiology and outcome of placental hypoxia, leading to a greater understanding of placental-mediated diseases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Part of this work was funded by the grant FONDECYT 1090355, Chile.