
Abstract
Abstract
Swenson, Erik R. Hypoxic pulmonary vasoconstriction. High Alt Med Biol 14:101–110, 2013.—Hypoxic pulmonary vasoconstriction (HPV) continues to fascinate cardiopulmonary physiologists and clinicians since its definitive description in 1946. Hypoxic vasoconstriction exists in all vertebrate gas exchanging organs. This fundamental response of the pulmonary vasculature in air breathing animals has relevance to successful fetal transition to air breathing at birth and as a mechanism of ventilation-perfusion matching in health and disease. It is a complex process intrinsic to the vascular smooth muscle, but with in vivo modulation by a host of factors including the vascular endothelium, erythrocytes, pulmonary innervation, circulating hormones and acid-base status to name only a few. This review will provide a broad overview of HPV and its mechansms and discuss the advantages and disadvantages of HPV in normal physiology, disease and high altitude.
Introduction
Characterization of Hypoxic Pulmonary Vasoconstriction
Increased pulmonary vascular resistance (PVR) and pulmonary artery (PA) pressure upon ascent to high altitude or exposure to normobaric hypoxia universally occur in humans and other mammals. HPV can be detected with elevations in altitude as low as 1600—2500 m or with reductions in FIO2 to 0.15–0.18 (Levine et al., 1997; Smith et al., 2012; Swenson et al., 1994). The magnitude of HPV can vary almost five-fold among individuals (Fig. 1; Gruenig et al., 2000) and among species, in part related to total pulmonary vascular smooth muscle (Fig. 2; Faraci et al., 1984; Tucker et al., 1975), and with time at altitude from minutes to several days (Dorrington et al., 1997; Groves et al., 1987). HPV is the earliest mechanism that elevates PA pressure and PVR with hypoxic or high altitude exposure, but ultimately other mechanisms (perhaps partly in reaction to the initial elevation of pressure initiated by HPV along with greater cardiac output), but also activation of pressure-independent hypoxia- sensitive inflammatory and proliferative pathways (Voelkel et al., 2013) may more importantly contribute to the sustained pulmonary vascular resistance as a consequence of vascular remodeling that is generally established within days to weeks of continuous alveolar hypoxia (Grover, 1985; Sommer et al., 2008). These aspects beyond early HPV are discussed elsewhere in this issue by Welsh and Peacock. Acute HPV progressively diminishes over time with sustained hypoxia in newcomers to high altitude, as assessed by a fall in pressure and resistance with oxygen breathing. As early as 8 hours (Dorrington et al., 1997) through 1–3 days (Kronenberg et al., 1971; Maggiorini et al., 2001), the rise in pressure cannot be quickly and fully reversed with return to normoxia (Fig. 2). Within one to several weeks there is little response to oxygen over the first several hours of inhalation (Dubowitz and Peacock, 2007; Groves et al., 1987; Rotta et al., 1956). Reversibility with oxygen is usually the method to assess acute HPV in subjects already at altitude rather than exposing the subjects to more hypoxia, which of course is the means to test for HPV at low altitude. Whether those that have little vasodilation with oxygen also have little further vasoconstriction with additional hypoxia has never been studied.
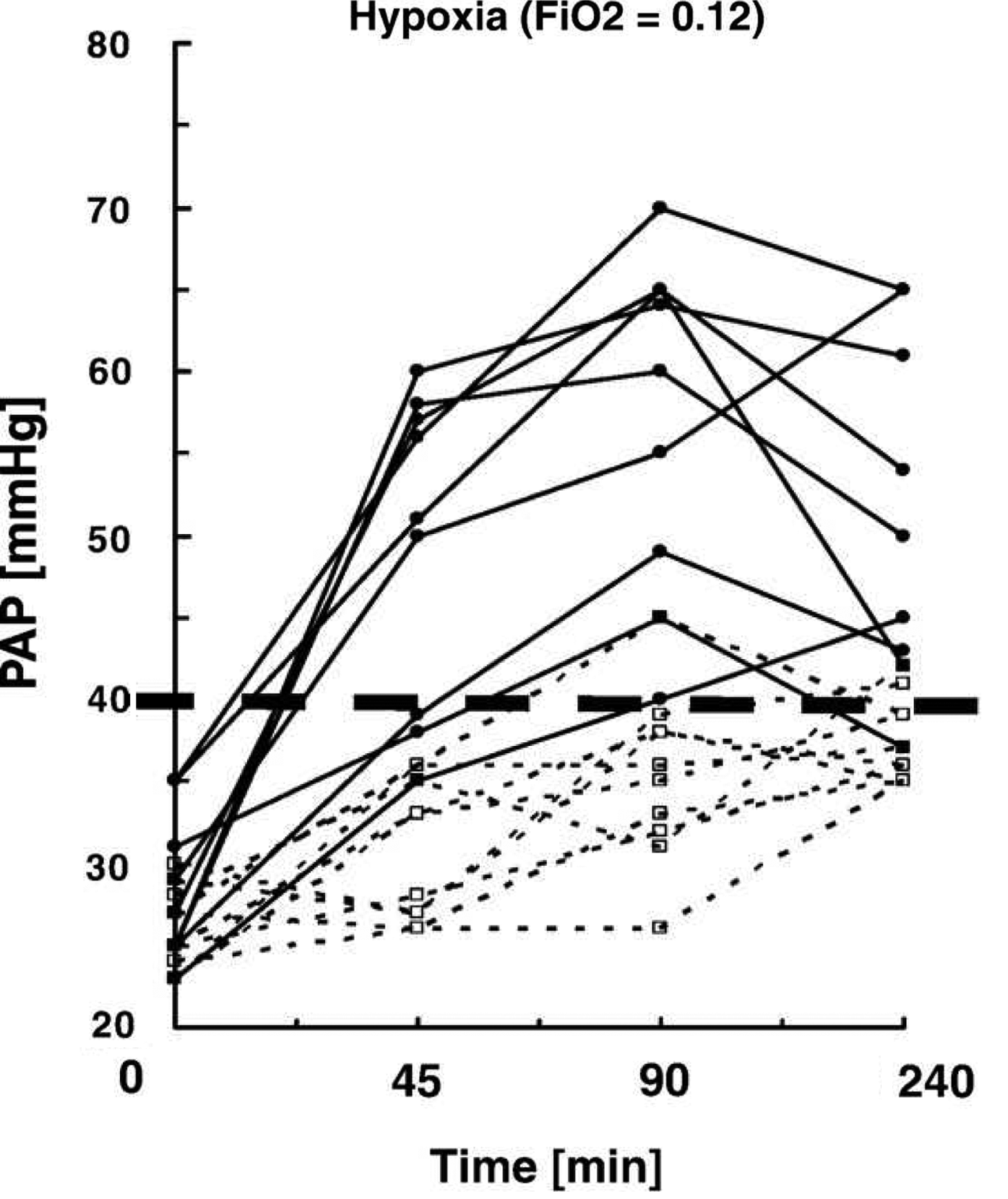
HPV variability as assessed by pulmonary artery systolic pressure response in normal humans to 4 h of moderate hypoxia. Subjects noted by solid lines are susceptible to high altitude pulmonary edema (HAPE) and show exaggerated HPV, while subjects without HAPE susceptibility (interrupted lines) have lower HPV (Grünig et al., 2000).
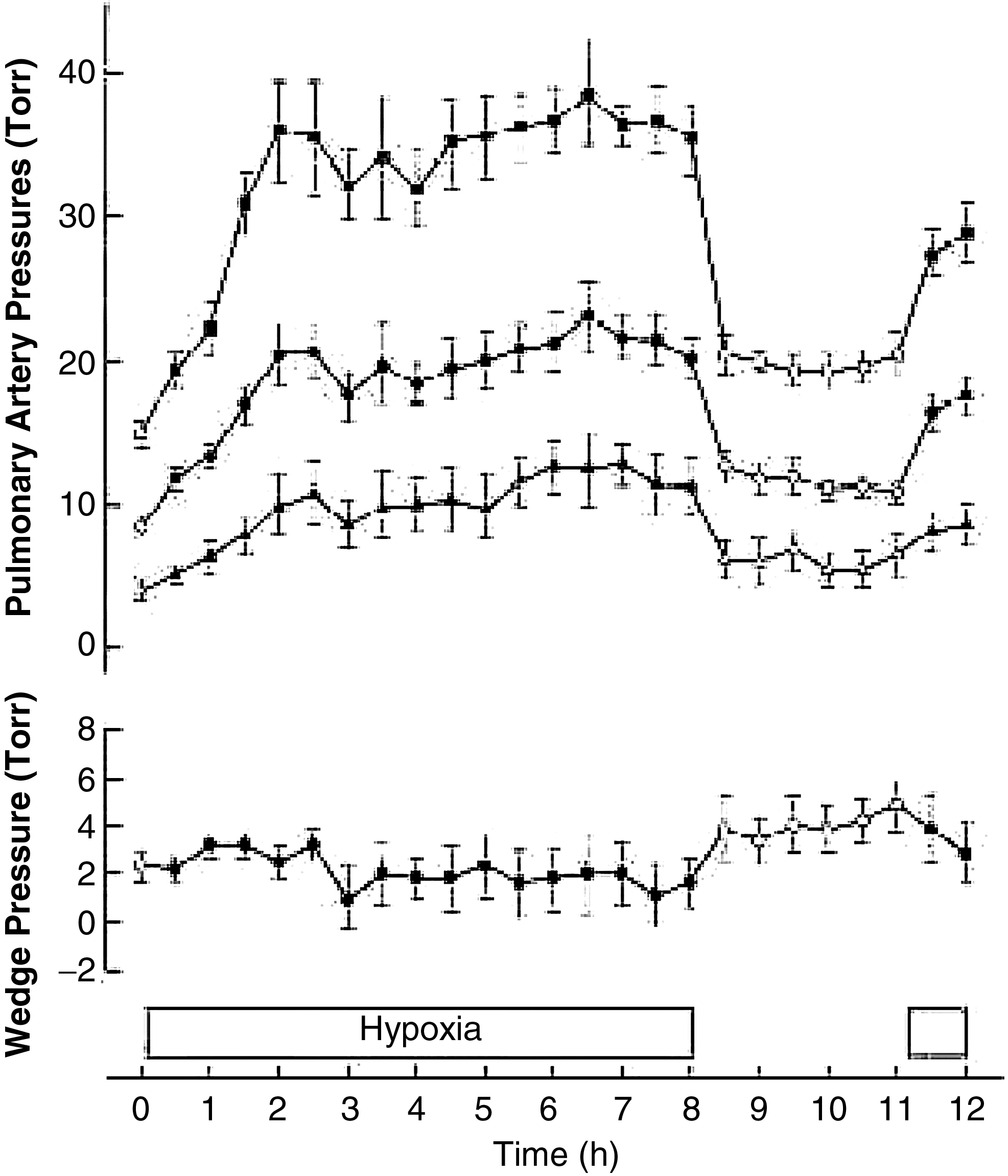
Time course in pulmonary artery pressure (systolic, mean, and diastolic) and wedge pressure in humans with acute hypoxia showing lack of complete resolution of HPV with return to normoxia after 8 hours (Dorrington et al., 1997).
Although changes in inspired oxygen remain widely used to assess HPV, it must be remembered that changes in arterial oxygenation and acid-base status always follow an alteration in inspired oxygen, so that systemic effects such as changes in CNS and autonomic nervous activity might also contribute to the pulmonary vascular response. It would be useful to employ a truly selective HPV inhibitor or stimulator in vivo rather than use changes in inspired oxygen, but all presently available pulmonary vasoactive agents have actions elsewhere in the circulation and brain, which make them less than ideal.
Lowland species with stronger acute HPV tend to develop greater pulmonary hypertension with chronic hypoxia than animals with weaker HPV (Figs. 3 and 4). In contrast, species that are native to high altitude have relatively low pulmonary artery pressures and blunted HPV (Durmowicz et al., 1993; Faraci et al., 1984; Harris et al., 1982; Llanos et al., 2007) and those human populations having resided longest at high altitude on the Tibetan plateau (Fig. 5) also have lower PA pressures (Groves et al., 1993; Hoit et al., 2005; Sun et al., 1993) and lesser HPV (Groves et al., 1993). These findings of reduced HPV after many generations at high altitude suggest either some element of eventual regression of remodeling or more likely environmental pressure that selectively favors individuals with low HPV.
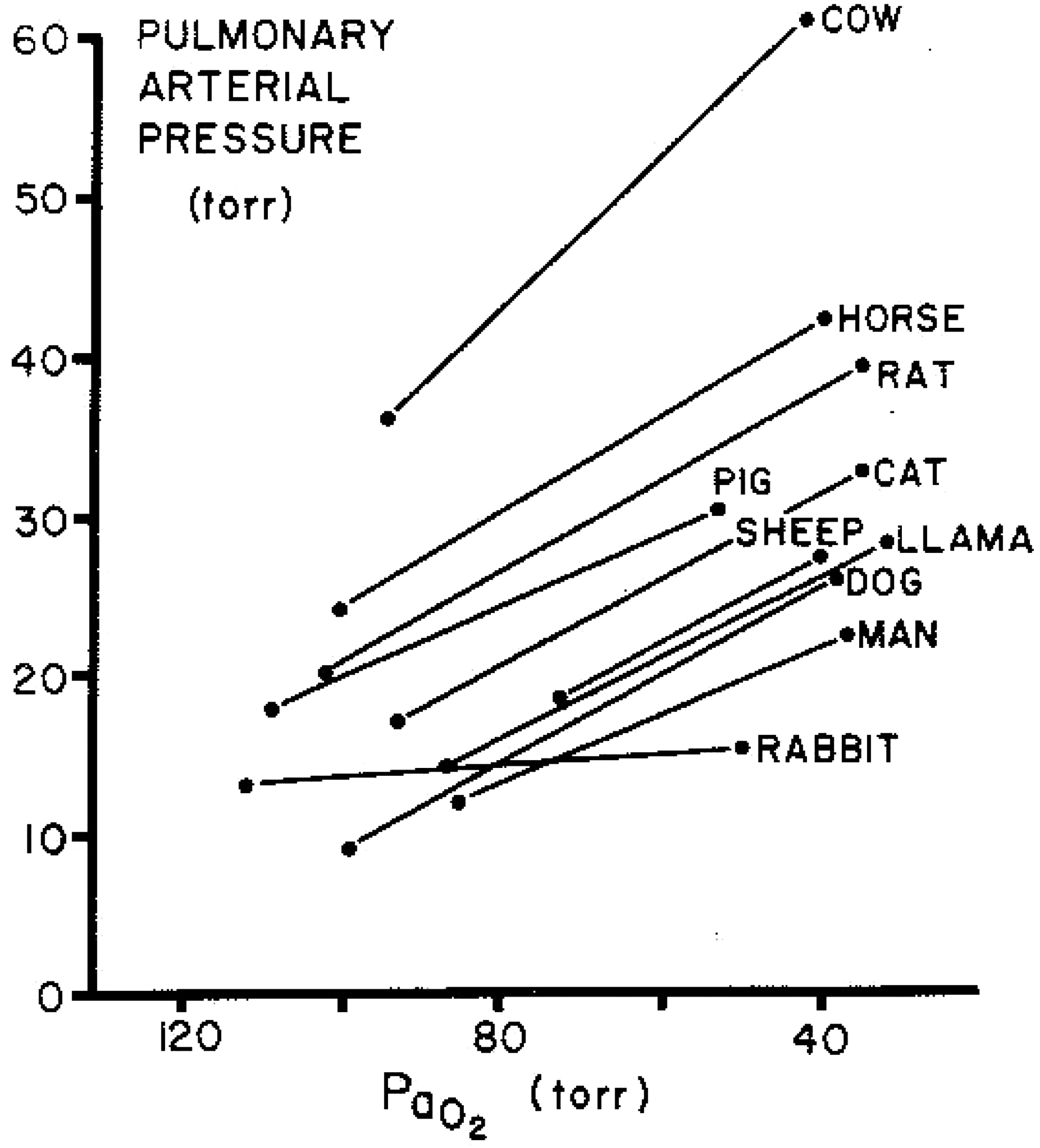
HPV in a variety of mammals showing baseline differences in normoxic mean pulmonary artery pressure and increases with acute hypoxia (Reeves et al., 1979).
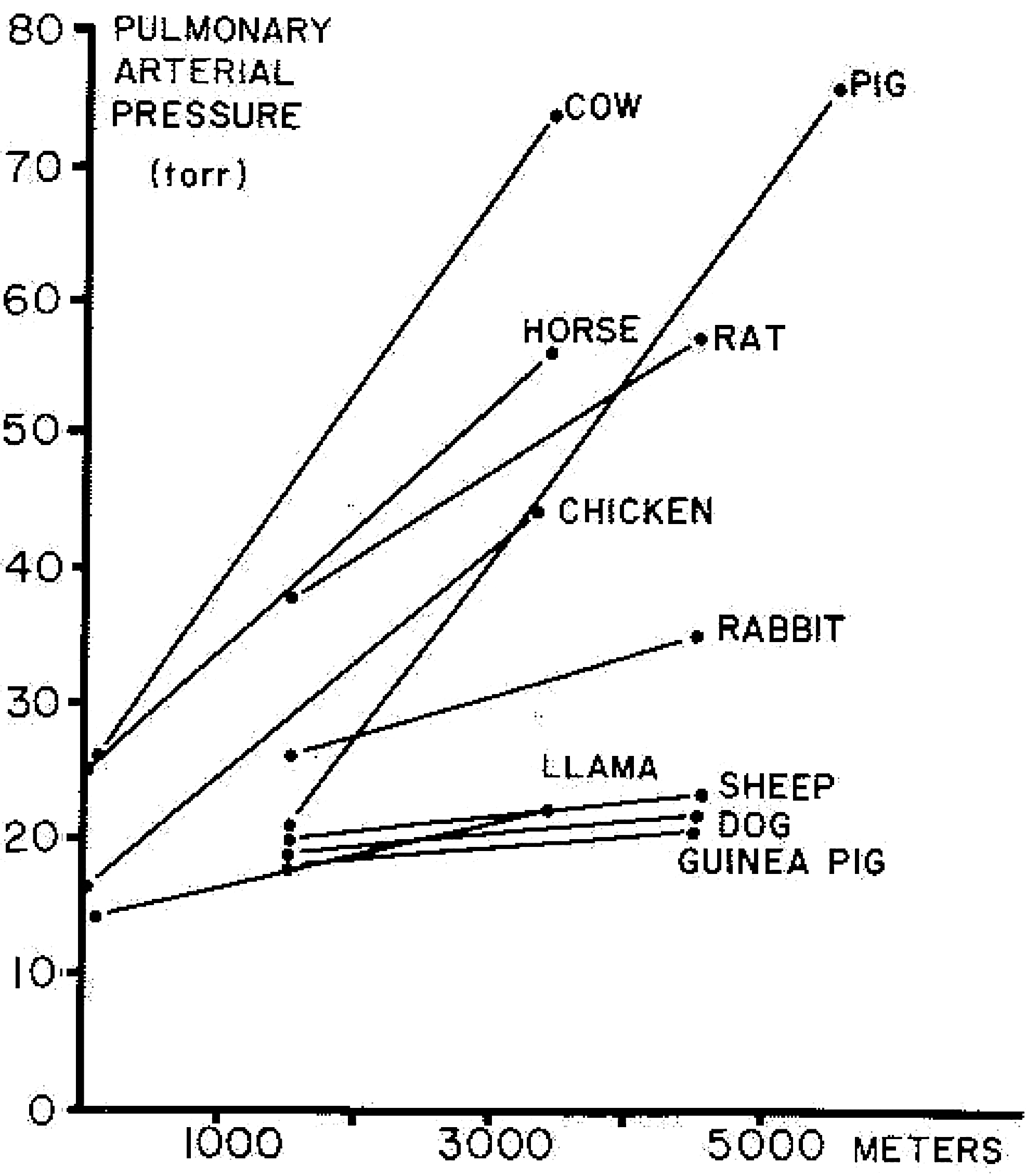
Species variability in severity of pulmonary hypertension during with chronic hypoxia (Reeves et al., 1979).

Mean pulmonary artery pressure at rest versus arterial P
The critical P
Mechanisms of Acute Hypoxic Pulmonary Vasoconstriction
HPV is a complex process with elements of its expression arising from multiple points in the neuro-cardiopulmonary axis, with variation in intensity and mechanisms over time (Sylvester et al., 2012). In addition to the intrinsic hypoxic response of pulmonary vessels that can be elicited in isolated pulmonary vascular smooth muscle cells and vessels, there are numerous extrinsic modulating influences sensitive to oxygen in vivo that include the vascular endothelium, red cells, chemoreceptors, autonomic nervous system, and lung innervation, as shown in Figure 6. The response of the pulmonary circulation to environmental hypoxia is characterized by contraction of smooth muscle cells within small pulmonary arterioles and veins of a diameter less than 900 μm, the veins accounting approximately for 20% of the total increase in pulmonary vascular resistance (Audi et al., 1991; Hakim et al., 1983). At a regional level within the lung vasculature, the magnitude of HPV may not be equivalent in all areas or static over time (Asadi et al., 2013; Dawson, 1969; Dehnert et al., 2006; Hopkins et al., 2005; Urano et al., 2005). As a consequence of this unevenness of regional HPV, some areas of the vasculature may be more perfused than others if they have a lower HPV response relative to other areas. This appears to be the case in those with a stronger global HPV response and susceptibility to high altitude pulmonary edema (Dehnert et al., 2006; Hopkins et al., 2005). Although it is not generally thought that hypoxia acts at the microvascular or acinar level, pulmonary capillary endothelial cells respond to hypoxia with membrane depolarization (Wang et al., 2012). As yet, no evidence has been found for capillary constriction (Conhaim et al., 2008) despite evidence that other vasoconstrictors are active at this level and in surrounding parenchymal perivascular cells that contain actin and myosin microfilaments (Watson et al., 2012).
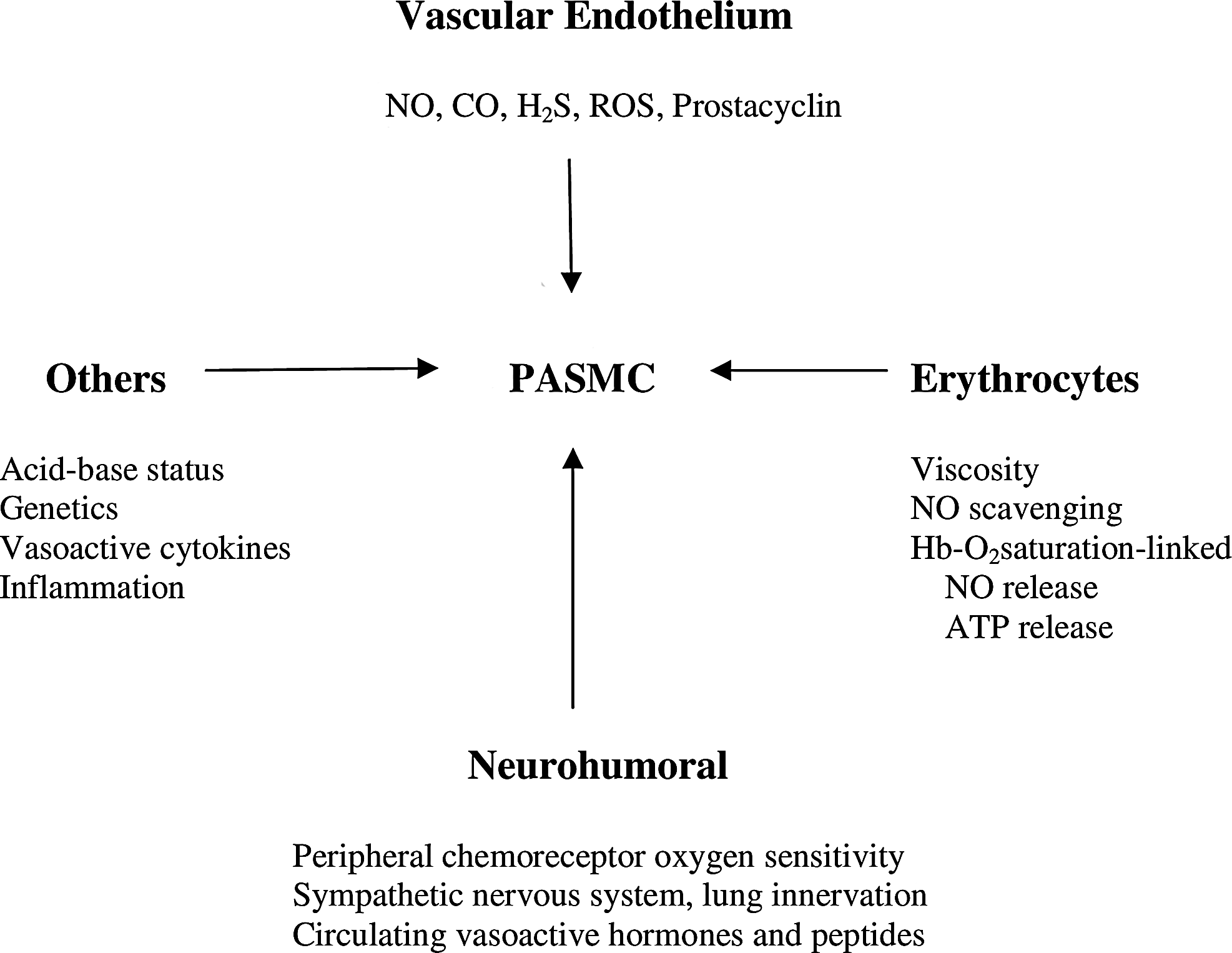
Diagram of modulating influences on HPV (see text for details).
HPV in intact animals and humans appears to be fully expressed within 6–8 hours and has several temporal components. The first occurs within 5 minutes with a half time of about 30–90 seconds (Deem et al., 2000; Johansen et al., 1998; Morrell et al., 1995; Talbot et al., 2005; Vejlstrup et al., 1997). A second phase of greater pressure elevation (almost double) is evident in humans and plateaus at 2 hours (Talbot et al., 2005). In animal studies, further elevation of pressure develops over the next 6–8 hours (Vejlstrup et al., 1997). This has been confirmed in studies of isolated pulmonary arteries, lungs, or vascular smooth muscle cells showing a third phase taking upward of 8 hours (Aaronson et al., 2002). Although isolated vessels or lungs appear to show a roll off in the increased pressure after initial exposure to hypoxia after 10–15 min which then subsequently rises again to greater levels over the next several hours (Aaronson et al., 2002), this has not been observed in vivo (Dorrington et al.1997; Teppema et al., 2007). The mechanisms behind these differing time phases and differences between in vivo and isolated lung and vessel investigations has not been well studied, but the isolated vessel studies suggest the first phase is intrinsic calcium-dependent smooth muscle contraction, with the later phases representing the summation of numerous other modulating influences acting on the smooth muscle (Aaronson et al., 2002; Sylvester et al., 2012) in a calcium concentration-independent fashion as discussed below. The absence of an early roll off in pressure in vivo but not in isolated lungs and vessels is not explained, but intact innervation of lung in vivo may play a role (see below). The roll off may be a phenomenon related to the much more severe hypoxia that can be applied in the isolated situations, or is the result of the increase in cardiac output that is not modeled in the isolated lung studies. All of these differing hypoxic responses are fully and immediately reversible with return to normoxia if hypoxia is not extended beyond several hours.
HPV at the level of the vascular smooth muscle
There are several mechanisms involved in HPV that are activated in parallel or sequentially, leading to the critical increase of intracellular calcium and/or an enhanced calcium sensitivity of the actin-myosin that initiates contraction (Sommer et al., 2008; Sylvester et al., 2012). Intracellular calcium concentration is increased by hypoxia-mediated inhibition of several potassium channels, leading to membrane depolarization and extracellular calcium entry through L-type channels, and a release of calcium from the sarcoplasmic reticulum (SR), with consequent further influx through store-operated calcium channels (SOCC), receptor-operated calcium channels (ROCC), and transient receptor potential channel 6 (TRPC6). Figure 7 depicts the very complicated and multiple pathways by which intracellular calcium in pulmonary vascular smooth muscle is quickly altered by hypoxia to initiate HPV. In addition, sensitivity to calcium of the contractile elements is enhanced via a hypoxia-induced increase in Rho-kinase activity (Weigand et al., 2011). The change in oxygen tension that stimulates these components of HPV is signaled by an alteration in the redox status of the smooth muscle cells (Schumacker, 2011; Sommer et al., 2008). Whether an increase or a decrease of reactive oxygen species (ROS) is responsible for HPV signal transduction is still under debate, but a stronger case is emerging that hypoxia increases mitochondrial ROS generation as an upstream signal for HPV (Schumacker, 2011). It is clear that high altitude exposure increases stable circulating markers of ROS production and persons with higher HPV appear to generate more ROS and less bioactive vasodilating NO species across the lung (Bailey et al., 2010).

Diagram of PASMC intracellular calcium ([Ca2+]i) pathways with acute hypoxia. Pathways that increase ([Ca2+]i are shown on the left, while those that decrease ([Ca2+]i are on the right. Hypoxia can activate (green) or inhibit (red) these pathways. Whether these effects are probable, possible, or speculative is indicated by solid, dashed, and dotted lines, respectively, as shown in the key at the bottom. With respect to plasma membrane and associated cytosolic signals, TASK-1 is TWIK-related acid-sensitive channel-1; VOCC, KV, ClCa, SOCC, NSCC, and ROCC indicate voltage-operated Ca2+, voltage-dependent K+, calcium-dependent Cl, store-operated Ca2+, nonselective cation, and receptor-operated Ca2+ channels, respectively. A, agonist; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; NCX, Na-Ca exchanger; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PLC, phospholipase C; PMCA, plasma membrane Ca2+-ATPase; R, receptor. With respect to sarcoplasmic reticulum (SR) and associated cytosolic signals: ADPR, cyclic ADP ribose; IP3R, IP3 receptor, RyR, ryanodine receptor, SERCA, sarcoplasmic-endoplasmic reticulum ATPase, STIM1, stromal interaction molecule 1. With respect to lysosome-like organelles (LLO) and associated cytosolic signals: HCX, H-Ca exchanger; HA, H+-ATPase; Mito, mitochondria; NAADP, nicotinic acid adenine dinucleotide phosphate (Sylvester et al., 2012).
Endothelium-dependent modulation of HPV
The pulmonary vascular endothelium generates a variety of vasoactive mediators that act in a paracrine fashion on the surrounding vascular smooth muscle cells. These include nitric oxide and prostacyclin as vasodilators, and endothelin-1 acting both as a vasoconstrictor via binding to endothelin A receptors and a vasodilator by binding to endothelin B receptors causing NO release (Aaronson et al., 2008). Isolated human PA endothelial cells exposed to 3% oxygen produce more hydrogen peroxide and thus may also be a source for ROS that initiate HPV (Irwin et al., 2009). The endothelium also produces carbon monoxide (CO) via heme-oxygenase-2 (Zhang et al., 2004), which is upregulated by hypoxia (Llanos et al., 2012; Motterlini et al., 2000). CO dilates vessels by activating guanylate cyclase to generate cyclic GMP in a manner similar to NO. Hydrogen sulfide (H2S), a strong reducing agent, generated in hypoxia, is vasoconstricting in the pulmonary circulation by several as yet not fully quantified mechanisms (Madden et al., 2012). It should be noted that many of these ‘gaso-transmitters' alter the concentrations of each other, making it difficult to assess the contribution of each to HPV modulation (Evans et al., 2011; Skovgaard et al., 2011).
As mentioned above, pulmonary microvascular endothelial cells respond to hypoxia with membrane depolarization. Wang et al. (2012) have suggested that the capillary vascular endothelial cells, owing to their intimate proximity to alveolar gas, are the site where rapid and efficient oxygen sensing occurs to initiate HPV. They propose that their membrane depolarization is propagated in a retrograde fashion to the upstream resistance vessels via inter-endothelial cell connexion 40-dependent gap junctions. The evidence for this novel concept is supported by lesser HPV, and greater VA/Q mismatching and hypoxemia when connexion 40 deficient mice are exposed to hypoxia or regional ventilation reduction.
Erythrocyte-dependent modulation of HPV
Red cells may contribute to HPV and pulmonary pressures in several ways. Although hypoxia-mediated decrease in deformability might reduce flow and increase measured vascular resistance (Hakim et al., 1988; Palareti et al., 1984), direct measurements of human and other mammalian red cells over a range of P
Neurohumoral-dependent modulation of HPV
The lung vasculature is innervated by sympathetic noradrenergic fibers from the large conduit arteries and veins down to 50 μm vessels in larger species such as man and dogs, but much less so in smaller species (Kummer, 2011). In addition to the release of norepinephrine with sympathetic activation causing vasoconstriction via alpha-1 adrenergic receptors on vascular smooth muscle, other opposing vasodilating neurotransmitters such as neuropeptide Y and vasoactive intestinal peptide can be released (Kummer, 2011). Additionally there is opposing vasodilating parasympathetic innervation that is NO dependent (McMahon et al, 1992). Arterial PO2 is gauged by the peripheral chemoreceptors, which then project afferents to the medullary cardiovascular control areas in the brain stem, in addition to the respiratory control center to activate both parasympathetic and sympathetic outflow to the lung. Denervation of the carotid bodies and loss of afferent input from the peripheral chemoreceptors increases HPV (Levitzky et al., 1978; Naeije et al., 1989). The efferent arc of this response is not well defined but is conveyed by the vagus nerve because vagotomy reduces HPV (Chapleau et al., 1988; Wilson et al., 1989). Studies using atropine and propranolol suggest that vasodilating parasympathetic activity is more dominant than sympathetic activity in HPV inhibition (Marshall, 1994; Wilson et al., 1989). Other data suggest a stronger sympathetic contribution (Lejeune et al., 1989). However, not all studies find evidence for neural modulation of HPV (Liu et al., 2007; Lodato et al., 1988). The reason for this discrepancy is not clear, but those studies finding no effect on HPV have employed receptor blocking drugs rather than neural pathway interruption. It is entirely possible that peripheral chemoreceptor-mediated modulation of HPV may involve other neurotransmitter release via the lung innervation besides catecholaminergic or cholinergic agonists as described above. In humans, the finding that a stronger hypoxic ventilatory response (HVR), which is almost wholly a peripheral chemoreceptor-mediated response, is associated with weaker HPV supports the majority of the animal work (Albert and Swenson, 2011). In the only other human study relevant to this issue and using a common Sp
Given that persons with exaggerated HPV are at greatest risk for high altitude pulmonary edema (HAPE) (Bartsch et al, 2005), it would be interesting to determine which of the many processes contributing to HPV are most important in this subset of vulnerable people. In regard to neurohumoral mediation of HPV, HAPE susceptibility may stem from greater generalized sympathetic nervous system activation to hypoxia (Duplain et al., 1999; Koyama et al., 1988) and lesser vascular endothelial nitric oxide production (Berger et al., 2005), at least as has been shown to be true in systemic organs, but not yet studied in the lung. Furthermore, owing to lesser hypoxic peripheral chemoreceptor responsiveness and lower ventilatory response to hypoxia (Hohenhaus et al., 1995; Selland et al., 1993) HAPE-susceptible subjects will breathe less at any given altitude or ambient PI
The pulmonary vasculature expresses adrenergic and cholinergic receptors, as well as other receptors, such as for thyroxine, angiotensin II, adenosine, natriuretic peptides, and estrogen, to name only a few. Thus it can respond to circulating vasoactive mediators with dilation by epinephrine via beta-2 receptors (Kummer, 2011), estrogens (Lahm et al., 2012), and natriuretic peptides (Cargill et al., 1996), and constriction with angiotensin (Hubloue et al. 2004), adenosine (Thomas and Marshall, 1993), and thyroxine (Herget et al., 1987). The full neuro-humoral response of the lung vascular response to hypoxia is often neglected in discussions of HPV, and while admittedly difficult to study, better understanding is much needed.
Other modulating influences on HPV
In addition to the many mechanisms involved in the control of pulmonary vascular tone in the hypoxic environment, individual genetic background (Kriemler et al., 2008; Leon-Velarde et al., 2008; Shirley et al., 2005; Stobdan et al., 2008), a history of familial susceptibility to HAPE or pulmonary hypertension (Grünig et al. 2010; Lorenzo et al., 2009; Scoggin et al., 1977), and environmental factors such as cold, intensity of exercise activity, and other stressors (Bartsch et al., 2005) also contribute to the strength of HPV. Acid-base status and carbon dioxide have a considerable influence on HPV, with alkalosis and hypocapnia both diminishing HPV in animals and humans (Balanos et al., 2003; Ketabchi et al., 2009), although changes in carbon dioxide without the usual accompanying changes in systemic pH may have opposite effects (Brimioulle et al., 1990). Thus subjects with stronger ventilatory responses to hypoxia will not only maintain higher alveolar P
Hypoxia-regulated gene transcription factors and HPV
The study of HPV continues to identify new sensing, signaling, and effector mechanisms and pathways. The most recent are the hypoxia-inducible factors (HIFs). In two rat strains with differing pulmonary hypoxic responses, HIF-1 activity and HIF-mediated protein expression were higher in the strain with greater pulmonary hypertension (Engebretsen et al., 2007). In contrast, mice with heterozygous HIF 1-alpha deficiency have weaker acute and chronic hypoxic responses in isolated pulmonary vascular smooth myocytes and pulmonary vessels than wild-type mice (Shimoda et al., 2001; Yu et al., 1999). Interestingly, carotid body sensitivity to hypoxia in these same HIF 1-alpha deficient heterozygote mice is depressed (Kline et al., 2002), although this does not appear to diminish the hypoxic ventilatory response. Further supporting pharmacological evidence for HIF-1alpha mediation of HPV was demonstrated in mice by reduction in hypoxic pulmonary hypertension (Abud et al., 2012) and increased ventilation in humans during hypoxic exercise (Janssen et al., 2010) with digoxin, a known inhibitor of HIF-1alpha transcriptional activity (Zhang et al., 2008). At present it is not fully clear how HIF-dependent gene transcription affects HPV, but it likely involves upregulation of transient receptor potential channels (TRPC) on the vascular smooth muscle cell membrane (Wang et al., 2006) and alterations in pulmonary vascular smooth muscle calcium signaling (Abud et al., 2012). Iron is emerging as a critical element in HPV and pulmonary vascular changes with hypoxia. Iron supplementation and iron chelation reduce and increase HPV respectively (Smith et al., 2008; Smith et al., 2009), possibly via altered HIF metabolism (Knowles et al., 2003) involving prolyl hydroxylases, the O2 sensitive enzymes that degrade HIF and require iron. HIF-mediated gene transcription also drives much of the longer term remodeling of the vasculature, as described by Welsh and Peacock in this issue.
Relevance of Hypoxic Pulmonary Vasoconstriction
At low altitudes where humans evolved, it would appear that the sensitivity to oxygen of the lung vasculature evolved along with hypercapnic pulmonary vasoconstriction (HCPV) as mechanisms (Dorrington et al., 2010) to shift blood flow from poorly or nonventilated lung regions with localized airway or airspace pathology in post-fetal life to better ventilated and healthy areas as elegantly advanced by von Euler and Liljestrand (1946) in their landmark paper. Based on whole lung pulmonary vascular responses to changes in alveolar P
Alternatively, others have argued it may be simply a vestige of fetal existence. In this regard, HPV maintains a high vascular resistance to limit blood flow in the nonventilated lung (in combination with a patent ductus arteriosis and foramen ovale) so as to allow a 80%–90% right to left shunt to provide more blood flow to the placenta and better oxygenated blood to the developing brain (Gao and Raj, 2010). However, many other aspects of the fetal lung also contribute to higher PVR including its liquid-filled nonventilated low volume state, lack of surfactant, relative hypercapnia and acidosis, a limited slower growing vascular bed relative to the faster growing airway and parenchymal structure, lesser endothelial vasodilator generation, greater endothelial vasoconstrictor production, and lack of bronchial epithelial NO generation (Gao and Raj, 2010; Morin et al. 1992). In fact, HPV in the fetal lung does not appear until the middle of the third trimester of gestation to reduce PVR and prepare the pulmonary circulation to accommodating the entire cardiac output at birth as the ductus arteriosis closes and the lungs are ventilated and assume gas exchange duties from the placenta (Morin et al, 1992). In this sense, HPV should perhaps more correctly be renamed ‘oxygen-dependent vasodilation’. If strong HPV is an evolutionary advantage in utero, then one might predict a fetal survival disadvantage in Tibetans, who have as adults much lower HPV than other populations (Groves et al., 1993). Yet, birth rates and neonatal survival in this population exceed those of newcomers to high altitude (Moore et al., 1998).
The physiologic advantage of HPV, if any, in healthy humans at high altitude appears nil. It has been postulated that moderately elevated pulmonary artery pressure optimizes systemic oxygen delivery by increasing blood flow into areas of lung with relatively lower blood flow, thus recruiting a greater fraction of the total alveolar capillary surface area for gas exchange (Wagner et al., 1979). Additionally this might be enhanced by hypoxic pulmonary venoconstriction as well (Taylor et al., 2011). HPV and the associated cardiac output increase at rest and with exercise does improve diffusing capacity by recruitment of alveolar capillary surface area for gas exchange and thus may improve arterial oxygenation (Steinacker et al., 1998). In the normal dog lung, hypoxia (equivalent to FI
Hypoxic pulmonary vasoconstriction can be increased or decreased for prophylactic and treatment purposes by a variety of pharmacological agents that act on many of the endothelial cell derived modulators of pulmonary vascular resistance, signal transduction pathways, and gene transcription discussed in detail elsewhere in this issue. Drugs which blunt or abolish HPV at high altitude are quite useful to prevent and treat high altitude pulmonary edema and high altitude pulmonary hypertension. These same drugs that inhibit HPV at low altitude, on the other hand, may impair gas exchange in a variety of settings of acute cardiac and lung injury and in chronic lung disease where their use is directed toward lowering systemic and/or pulmonary artery pressure or in increasing exercise capacity (Blanco et al., 2010; Cutaia and Rounds, 1990; Preston, 2007). Under some situations, medications that enhance HPV can be utilized to improve gas exchange particularly during cardiothoracic surgery (Nagendran et al., 2006). The search for more potent and selective vasodilators for the treatment of nonhypoxic forms of pulmonary hypertension grows apace, and it is likely that most will have the ability to inhibit HPV both for clinical purposes and as an unwanted side effect depending upon the clinical circumstances.
Author Disclosure Statement
No competing financial interests exist.