
Abstract
Brewster, L. Madden, Anthony R. Bain, Vinicius P. Garcia, Noah M. DeSouza, Michael M. Tymko, Jared J. Greiner, and Philip N. Ainslie. Global REACH 2018: high altitude-related circulating extracellular microvesicles promote a proinflammatory endothelial phenotype in vitro. High Alt Med Biol. 24:223–229, 2023.
Introduction:
Ascent to high altitude (HA) can induce vascular dysfunction by promoting a proinflammatory endothelial phenotype. Circulating microvesicles (MVs) can mediate the vascular endothelium and inflammation. It is unclear whether HA-related MVs are associated with endothelial inflammation.
Objectives:
We tested the hypothesis that MVs derived from ascent to HA induce a proinflammatory endothelial phenotype.
Methods:
Ten healthy adults (8 M/2 F; age: 28 ± 2 years) residing at sea level (SL) were studied before and 4–6 days after rapid ascent to HA (4,300 m). MVs were isolated and enumerated from plasma by centrifugation and flow cytometry. Human umbilical vein endothelial cells were treated with MVs collected from each subject at SL (MV-SL) and at HA (MV-HA).
Results:
Circulating MV number significantly increased at HA (26,637 ± 3,315 vs. 19,388 ± 1,699). Although intracellular expression of total nuclear factor kappa beta (NF-κB; 83.4 ± 6.7 arbitrary units [AU] vs. 90.2 ± 6.9 AU) was not affected, MV-HA resulted in ∼55% higher (p < 0.05) active NF-κB (129.6 ± 19.8 AU vs. 90.7 ± 10.5 AU) expression compared with MV-SL. In addition, MV-HA induced higher interleukin (IL)-6 (63.9 ± 3.9 pg/ml vs. 53.3 ± 3.6 pg/ml) and IL-8 (140.2 ± 3.6 pg/ml vs. 120.7 ± 3.8 pg/ml) release compared with MV-SL, which was blunted with NF-κB blockade.
Conclusions:
Circulating extracellular MVs increase at HA and induce endothelial inflammation, potentially contributing to altitude-related vascular dysfunction.
Introduction
Ascent to high altitude (HA; >2,400 m) increases the risk of vascular conditions, such as endothelial dysfunction, cerebral and pulmonary edema, and pulmonary hypertension (Bailey et al., 2009; Berger et al., 2005; Luks and Swenson, 2007; Luks et al., 2019; Luks et al., 2017). Although unclear whether inflammation is an essential cause/cofactor or consequence of these HA-related pathologies, changes in vascular endothelial cell function, most notably the development of a proinflammatory endothelial phenotype, have been shown to play a role (Hartmann et al., 2000; Himadri et al., 2010; Julian et al., 2011; Mishra et al., 2016; Song et al., 2016; Swenson et al., 2002).
Indeed, systemic inflammation and acute hypoxia have been shown to elicit cerebral edema in rats, indicating that inflammation may play a role in the onset of HA cerebral edema (Song et al., 2016). Despite incongruencies in local versus systemic inflammation as well as etiological cause for pathology, it is clear that altitude exposure is associated with inflammation; however, the mechanism(s) driving this acute HA-related vascular inflammation remain unclear.
The vascular endothelium is significantly influenced by circulating cell-derived microvesicles (MVs) (Jansen et al., 2017; Koganti et al., 2017). MVs are small (100–1,000 nm in diameter) extracellular vesicles that bud from the plasma membrane of various cell types in response to a myriad of physiologic and pathologic stimuli, including hypoxia (Ratajczak and Ratajczak, 2020; Tremblay et al., 2017; Venturella et al., 2021).
Circulating MVs can have profound effects on systemic responses such as inflammation and can also functionally affect specific tissues (Hijmans et al., 2019; Słomka et al., 2018; Wahlund et al., 2017). For example, our laboratory and others have previously demonstrated that circulating MVs promote a proinflammatory endothelial phenotype (Brewster et al., 2021; Heinrich et al., 2015; Hijmans et al., 2019; Jansen et al., 2013). Moreover, MVs have previously been shown to be elevated with ascent to HA (Tremblay et al., 2017). It is currently unknown, however, if exposure to HA MVs induces endothelial inflammation. If so, MVs may provide an underlying mechanism by which HA drives vascular inflammation, contributing to the onset of HA-related pathology and conditions.
Accordingly, the experimental aim of this study was to determine the effect of acute HA-related MVs on endothelial cell inflammation. We hypothesized that MVs derived from ascent to HA induce a proinflammatory endothelial phenotype. To address this aim, we determined, in vitro, the effect of MVs isolated from healthy adults before and after ascent to HA on the key inflammatory transcription factor, nuclear factor kappa beta (NF-κB), and associated cytokine production.
Methods
Subjects
Ten lowlanders (8 M/2 F) from the Global Research Expedition on Altitude Related Chronic Health (REACH) 2018 expedition team participated in this study (Tymko et al., 2021). All subjects were free of overt cardiometabolic disease, permanently resided at or near sea level (SL), and did not travel to HA within 6 months before the study. The experimental protocols were approved by ethics boards at the University of British Columbia (H17-02687 and H18-01404) and Universidad Peruana Cayetano Heredia (No. 101686). All subjects had the potential risks and benefits of the study explained before providing written informed consent.
Experimental overview
Baseline measures were performed at the University of British Columbia (Kelowna, British Columbia, Canada, 344 m) 2 weeks before departure to Lima, Peru. Subjects flew into Lima, Peru, and shortly thereafter, ascended to HA (Cerro de Pasco, Peru, 4,300 m) by automobile (∼8 hours). Follow-up measures were performed after 4–6 days at HA.
Blood gases
Hemoglobin (Hb) and blood gasses (arterial oxygen saturation [SaO2]; partial pressure of arterial carbon dioxide [PaCO2]; and partial pressure of arterial oxygen [PaO2]) were analyzed using a commercial blood gas analyzer: ABL90 Flex blood analyzer (Radiometer, Copenhagen, Denmark). Plasma concentrations of inflammatory cytokines, interleukin (IL)-6 and IL-8, were determined by enzyme immunoassay (R&D Systems, Minneapolis, MN).
MV isolation and enumeration
Detailed descriptions of MV isolation and enumeration are provided elsewhere (Brewster et al., 2021; Brewster et al., 2020; Hijmans et al., 2019). In short, MVs were isolated through ultracentrifugation from platelet-free plasma and quantified by flow cytometry on a FACSAria I flow cytometer (BD Biosciences). MV size threshold was established using Megamix-Plus SSC calibrator beads (Biocytex, Marseille, France) and only events >0.16 and <1 μm in size were counted. These methods were in accordance with the most current guidelines set by the International Society for Extracellular Vesicles available at the time of our study (Lötvall et al., 2014).
Endothelial cell culture and MV treatment
Human umbilical vein endothelial cells (HUVECs; Life Technologies, ThermoFisher, Waltham, MA) were cultured under standard culture conditions as previously described by our laboratory (Brewster et al., 2021; Brewster et al., 2020; Hijmans et al., 2019). HUVECs were separately treated with MV (2e6 MVs:1e6 HUVECs) ratio from each individual subject isolated at SL (MV-SL) and HA (MV-HA) for 24 hours. In separate experiments, HUVECs were co-treated with 200 nM IκB kinase inhibitor (IKK-16) and MVs for 24 hours (Brewster et al., 2021). After 24 hours of incubation, the supernatant was collected for the determination of cytokine release and cells were harvested for the determination of cellular protein and microRNA (miRNA) expression.
Endothelial cell cytokine release
Concentrations of IL-6 and IL-8 in the HUVEC supernatant were determined using chemiluminescent immunoassay (R&D Systems). Intra-assay coefficient of variation for the media-based enzyme-linked immunosorbent assays was <10%.
Endothelial cell protein expression
Detailed methodology of the quantification of intracellular protein has been described previously (Brewster et al., 2021; Brewster et al., 2020; Hijmans et al., 2019). After 24 hours of treatment with MVs, HUVECs were washed in phosphate-buffered saline and protein extraction using RIPA lysis buffer supplemented with protease and phosphatase inhibitors, followed by sonication, and mechanical perturbation was performed. Protein concentration of lysates was determined by Bio-Rad DC protein assay (Bio-Rad, Hercules, CA). Protein expression was measured by capillary electrophoresis immunoassay using the Wes system (Wes; ProteinSimple, San Clara, CA). Rabbit primary antibodies against nuclear factor-κB p65 (NF-κB) (D14E12) and phospho-nuclear factor-κB (p-NF-κB) p65 (Ser536) (93H1) (both diluted 1:250) (Cell Signaling Technologies, Danvers, MA) were used.
Endothelial cell miRNA expression
Cellular expression of miR-146a and miR-181b was determined by reverse transcription polymerase chain reaction (RT-PCR) as previously described (Brewster et al., 2021; Brewster et al., 2020; Hijmans et al., 2019). Total cellular RNA was isolated using the miRVANA RNA isolation kit (Exiqon, Vedbaek, Denmark) and RNA concentration was determined using a Nanodrop Lite (ThermoFisher). RNA was reverse transcribed using the miScript II Reverse Transcription Kit (Qiagen, Hilden, Germany). RT-PCR was performed using the BioRad CFX96 RT-PCR platform with the miScript SYBR green PCR kit (Qiagen) using specific primers miR-146a, miR-181b, and U6 (Qiagen). miRNA expression was quantified using the comparative Ct method and normalized to U6. The relative expression of each transcript was calculated as the 2−ΔΔCt where 2−(Ct[miR] − Ct[RNU6]) and presented as arbitrary units (AU).
Statistical analysis
Normal distribution and homogeneity of variances were determined by the Shapiro–Wilk and Levene tests, respectively. Differences between repeated measures were made with the Student's paired t-test (normally distributed) or Wilcoxon test (non-normally distributed). Cellular cytokine release and protein expression were determined by repeated measures analysis of variance (ANOVA) (for normally distributed data) or Friedman test (for non-normally distributed data) followed by the Student-Newman-Keuls post hoc test, where applicable. Data in the text are presented as mean ± standard error of the mean (SEM) for normally distributed variables and as median (interquartile range [IQR]) for non-normally distributed variables. Statistical significance was set a priori at p < 0.05.
Results
Select subject characteristics are presented in the Table 1. Ascent to HA significantly increased blood pressure (systolic, p = 0.0147; diastolic, p = 0.0048) as well as Hb (p = 0.002). SaO2 (p = 0.0007), PaCO2 (p < 0.0001), and PaO2 (p < 0.0001) were all significantly lower at HA compared with SL. Circulating MVs (p = 0.0371) and plasma concentrations of IL-6 (p = 0.012) and IL-8 (p = 0.006) were higher at HA compared with SL.
Select Subject Characteristics
Due to technical difficulties, Hb (data: n = 8), SaO2, PaCO2, and PaO2 (data: n = 7) were not measured in all subjects. Values are mean ± SEM.
p < 0.05 versus sea level.
BMI, body mass index; BP, blood pressure; Hb, hemoglobin; IL-6, interleukin-6; IL-8, inteleukin-8; MV, microvesicle; PaCO2, partial pressure of arterial carbon dioxide; PaO2, partial pressure of arterial oxygen; SaO2, arterial oxygen saturation; SEM, standard error of the mean.
Endothelial cell inflammation
Effects of MVs on HUVEC cytokine release and intracellular protein expression are shown in Figure 1. Although intracellular protein expression of NF-κB p65 (mean ± SEM: 90.2 ± 6.9 AU vs. 86.7 ± 6.0 AU; p > 0.05) was not different between groups, cellular expression of p-NF-κB p65 (Ser536; active NF-κB) was ∼45% higher (mean ± SEM: 129.6 ± 19.8 AU vs. 83.9 ± 9.7 AU; p = 0.05) in HUVECs treated with MV-HA compared with MV-SL. HUVECs treated with MV-HA induced greater release of endothelial cell IL-6 (median [IQR]: 58.0 [54.0–59.9] pg/ml vs. 68.7 [64.3–60.7] pg/ml; p = 0.01) and IL-8 (median [IQR]: 123.8 [110.5–131.1] pg/ml vs. 138.7 [134.0–144.3] pg/ml; p = 0.0026) compared with MV-SL.
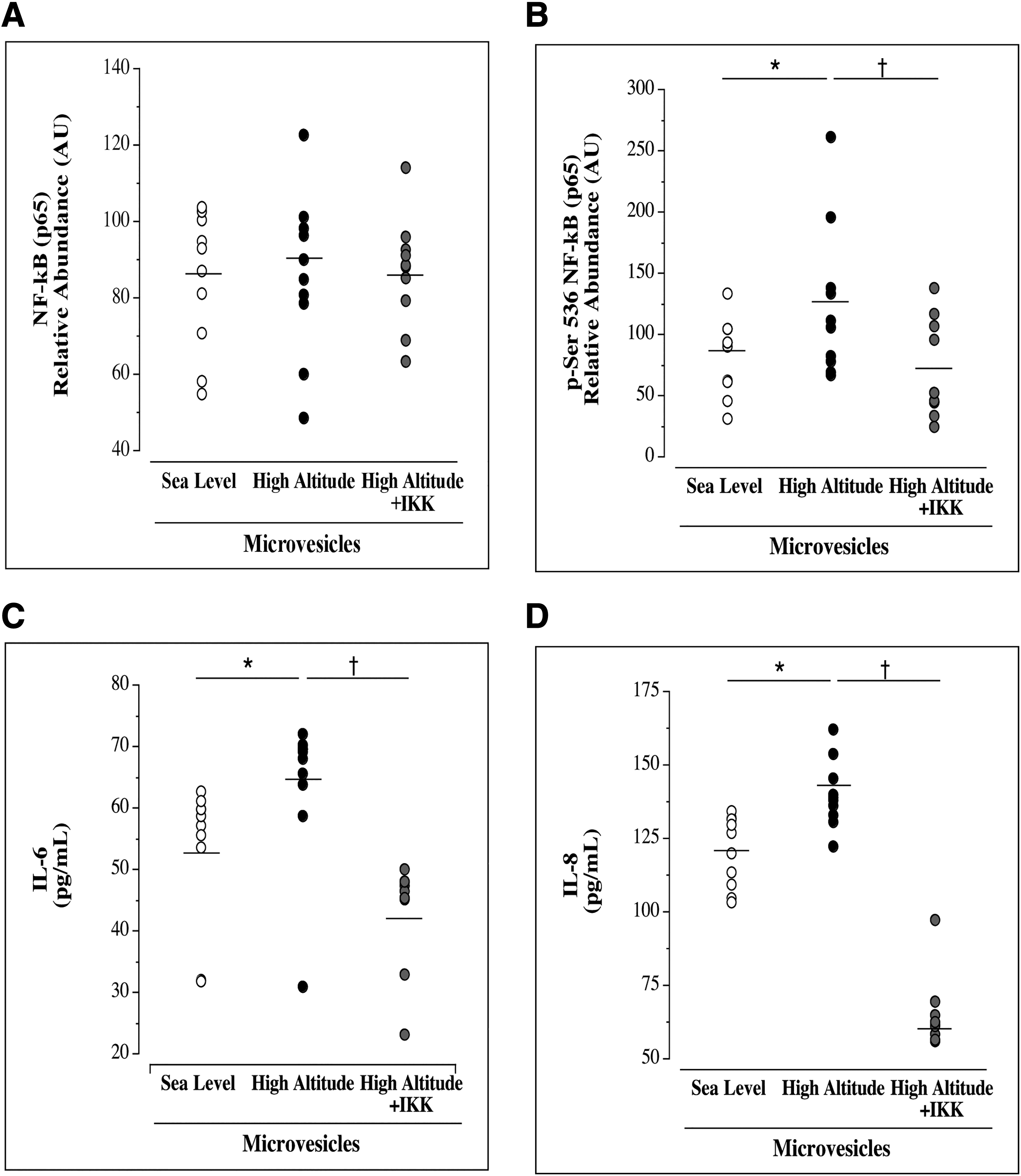
Intracellular protein expression of total NF-κB p65
An IKK complex inhibitor (IKK-16) was used to block the activation of NF-κB when co-treated with MV-HA. IKK-16 had no effect on total NF-κB p65 expression (mean ± SEM: 90.2 ± 6.9 AU vs. 87.3 ± 4.7 AU; p > 0.05); however, p-NF-κB (Ser536) expression (mean ± SEM: 129.6 ± 19.8 AU vs. 72.2 ± 12.9 AU) was ∼60% lower (p < 0.007) in cells co-treated with MV-HA and IKK-16 (Fig. 1). IKK-16 significantly blunted the effects of MV-HA on release of both IL-6 (median [IQR]: 68.7 [64.3–69.7] vs. 46.1 [45.3–47.9]; p = 0.0001) and IL-8 (median [IQR]: 138.7 [134.0–144.3] vs. 61.6 [57.3–64.5]; p < 0.0001).
Intracellular expression of miR-146a and miR-181b is shown in Figure 2. Intracellular expression of miR-146 (mean ± SEM: 1.7 ± 0.01 AU vs. 4.5 ± 0.05 AU; p = 0.0002) and miR-181b (mean ± SEM: 1.5 ± 0.01 AU vs. 4.2 ± 0.07 AU, p = 0.0023) was ∼60% and 65% lower, respectively, in HUVECs treated with MV-HA compared with MV-SL.

Endothelial cell expression of miR-146a
Discussion
The primary novel finding of this study is that MV-HA promotes endothelial cell inflammation. Herein, we demonstrate that MVs isolated from otherwise healthy adults who ascended to HA from SL increase endothelial cell activation of NF-κB. The activation of NF-κB was reaffirmed by enhanced release of IL-6 and IL-8 in endothelial cells treated with MV-HA compared with MV-SL. Moreover, key miRNAs associated with suppressing NF-κB activation were lower in cells treated with MV-HA. To our knowledge, this is the first study to determine the potential role of MVs in mediating vascular inflammation with rapid ascent to HA.
Hypoxia-inducible factors (HIF) are transcription factors that are activated by hypoxia (HA) and modulate a variety of physiological processes (Semenza, 2012). HIF-1α has previously been shown to elicit extracellular vesicle formation and release, particularly from the endothelium (Burnley-Hall et al., 2017; Wang et al., 2014). It is possible that the increase in total concentration of MVs observed in response to HA in this study is elicited by an increase in the endothelial cell-derived fraction of MVs in the total MV population. Moreover, HIF maintains essential crosstalk with NF-κB and thereby the inflammation pathway (Pham et al., 2021). The activation of HIF-1α and potential contribution to total and endothelial cell-derived MV release and the inflammatory response to HA should be addressed in future studies and may explain current observations.
NF-κB is the primary transcription factor responsible for regulating important inflammatory processes such as endothelial cytokine production (Brasier, 2010; Elliott et al., 2001). NF-κB transcription can be stimulated by a multitude of catalysts, including hypoxia; however, the mechanism(s) remain unclear (Culver et al., 2010; D'Ignazio et al., 2016; Oliver et al., 2009). We observed that MV-HA resulted in significantly increased phosphorylation of NF-κB compared with MV-SL.
The activation of NF-κB is associated with endothelial dysfunction and myriad of vascular diseases and conditions (Kempe et al., 2005; Pierce et al., 2009; Saito et al., 2013). One of the primary responses to endothelial NF-κB is the production and release of proinflammatory cytokines, such as IL-6 and IL-8. Given their culminating role in the NF-κB signaling cascade, IL-6 and IL-8 are also well-established markers of endothelial inflammation and circulating concentrations have previously been associated with HA exposure (Apostolakis et al., 2009, p. 6; Boos et al., 2016; Hartmann et al., 2000; Held et al., 2017, p. 6; Julian et al., 2011; Lundeberg et al., 2018; Mazzeo et al., 2001; Schieffer et al., 2004, p. 8).
To confirm that MV-induced NF-κB activation was directly responsible for changes in IL-6 and IL-8, endothelial cells were co-treated with the competitive IKK complex inhibitor, IKK-16. Specifically, IKK-16 blocks IKK complex phosphorylation of IκBα, the inhibitory subunit responsible for constitutively sequestering inactive NF-κB in the cytoplasm (Waelchli et al., 2006). While IKK-16 did not affect total intracellular NF-κB expression, MV-HA-induced increases in phosphorylated NF-κB were attenuated, confirming that IKK-16 inhibited NF-κB activation. Concomitantly, endothelial cell release of IL-6 and IL-8 was significantly blunted when NF-κB activation was blocked with MV-HA treatment. These findings indicate that MV-HA-induced changes in cellular release of proinflammatory cytokines, IL-6 and IL-8, are directly linked to NF-κB activation. As such, phosphorylation of NF-κB is responsible for the proinflammatory endothelial response elicited by MV-HA, thus providing novel mechanistic insight into acute attitude-related systemic inflammation.
Changes in NF-κB transcription by MVs may, at least in part, be due to changes in key miRNAs involved in NF-κB regulation. miRNAs are ∼22-nucleotide, noncoding, post-transcriptional regulators of protein expression and activity (Romaine et al., 2015). For example, miR-146a and miR-181b are two miRNAs known to regulate NF-κB activation (Habibi et al., 2016; Meng et al., 2020; Sun et al., 2014; Sun et al., 2012; Taganov et al., 2006). Specifically, miR-146a suppresses the initiation of NF-κB activation through adaptor proteins, IRAK1 and TRAF6, and miR-181b inhibits nuclear transport of NF-κB through importin-α-3 (Muroi and Tanamoto, 2008; Sun et al., 2012; Taganov et al., 2006). Indeed, MV-HA resulted in lower cellular expression of both miR-146a and miR-181b compared with MV-SL.
There are several limitations of this study. First, MV characterization was not confirmed following ultracentrifugation in this study, so it cannot be discounted that the effects demonstrated herein are from MVs alone and may be, in part, due to cellular debris and other extracellular vesicle fractions. Future studies are warranted to confirm MV characterization using updated guidelines. Second, sex-specific differences in response to HA exposure are limited and circulating MVs have been shown to differ between males and females (Bammert et al., 2017; Gustafson et al., 2015; Hou et al., 2019; Hunter et al., 2019). Therefore, possible sex-related differences in response to HA cannot be discounted. Due to limited female participants, future studies are needed to address this gap in the literature. Third, given that severity and duration of exposure significantly influence physiological outcomes at HA and we only included one time point at HA, future studies are needed to determine how MV number and function may change with these variables (Tymko et al., 2019).
In conclusion, the results of this study indicate that ascent to HA is associated with a proinflammatory circulating extracellular MV milieu. Indeed, MV-induced exacerbation of endothelial NF-κB activation and the associated increase in IL-6 and IL-8 production may underlie the propensity for HA-induced vascular inflammation. Future studies are warranted to extend our findings in the context of altitude illness.
Footnotes
Acknowledgments
We thank all of the subjects who participated in this study, as well as the staff at the University of Colorado Anschutz Medical Campus ACI/ID Flow Core for technical assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported, at least in part, by the Natural Sciences and Engineering Research Council of Canada (to Philip N. Ainslie) and Canada Research Chairs program (to Philip N. Ainslie).