
Abstract
As an issue of biosecurity, it is important to identify the origin of a suspected sample to distinguish whether it originated from the release of a bioterrorism agent or from environmental contamination with a virulent agent. Here we have developed an analytical pipeline that can infer the phylogenetic position of Bacillus cereus group species, including B. anthracis, from next-generation sequencing reads without extensive genomics skills. GcoGSA-BA can also detect the existence of anthrax plasmid pXO1 carrying 3 anthrax toxins (lethal factor, edema factor, and protective antigen). This pipeline can also be used to correctly infer the phylogenetic position and to detect the suspected isolate carrying anthrax toxins among B. cereus group.
It is important to identify the origin of a suspected sample to distinguish whether it originated from the release of a bioterrorism agent or from environmental contamination with a virulent agent. Here the authors have developed an analytical pipeline that can infer the phylogenetic position of Bacillus cereus group species, including B. anthracis, from next-generation sequencing reads without extensive genomics skills.
A
To conduct phylogenetic analysis of B. cereus group species (including B. anthracis) more conveniently, we implemented a web service designated as
Materials and Methods
The GcoGSA-BA web tool consists of simple pipelines that include read quality trimming, reference genome and toxin gene mapping, and phylogeny extraction based on the core genome sequence. Read trimming is performed using the skewer program 7 with a quality threshold of 15 and a minimum remaining sequence length of at least 40. The remaining reads are mapped, using the bwa-sw program, 8 to the reference genome sequences of B. anthracis “Ames Ancestor” 6 chromosome (NC_007530.2), including the 2 plasmids pXO1 (NC_007322.2) and pXO2 (NC_007323.3), which contain genes associated with toxin and capsule synthesis, respectively. 6 The number of mapped reads, the covered region, and the average read depth for the reference sequences are estimated from the mapping result. Reliable SNP sites are selected from the mapped sites with at least 5×coverage depth, and those of the 3 plasmid-coded toxin proteins (LF, EF, and PA) are estimated separately.
A total of 657,183 precomputed SNP sites were extracted from 29 B. anthracis and 104 B. cereus strains (https://genome.niid.go.jp/gcogsa/images/bacillus/SNP-allele.table.gz). The SimSeq program was employed to generate the simulated reads from the complete or draft genome sequence of the above Bacillus strains (SimSeq available at https://github.com/jstjohn/SimSeq). A neighbor-joining (NJ) phylogenetic tree 9 could then be constructed from the concatenated 657,183 SNP alleles using the APE package of R. 10 A zoomed-in version of the phylogenetic tree surrounding the query strain(s) is also created for user convenience. The results can then be downloaded from the website, including the SNP table in tabular format and multifasta format, NJ phylogenetic tree in Newick format and PDF format, and mapped ratios for further analysis.
The source code for installing on a local server is available on request. The GcoGSA-BA website is https://genome.niid.go.jp/gcogsa/BA/index.html.
Results and Discussion
In total, 657,183 SNP sites on B. anthracis Ames Ancestor chromosome DNA (5,227,419 bp) were extracted using complete or draft genome sequence from 29 B. anthracis and 104 B. cereus strains, indicating that 12.57% of chromosome regions were comprehensively selected for phylocore-genome analysis. To evaluate the performance of GcoGSA-BA, we challenged raw or simulated data obtained from 4 types of Bacilli: 2 strains of B. anthracis (Ames Ancestor and A0149) and 2 strains of B. cereus (G9241 and VD166). Sequence read archive data for B. anthracis A0149 and B. cereus VD166 were retrieved from SRR1318540 and SRR398308 in NCBI (http://www.ncbi.nlm.nih.gov/Traces/sra), respectively, and simulated short reads were prepared from B. anthracis Ames Ancestor (chromosome DNA, NC_007530.2; pXO1, NC_007322.2; pXO2, NC_007323.3) and the draft genome assembly of B. cereus G9241 (AAEK01000001-AAEK01000207). All of the tested samples were deeply mapped to the reference Ames Ancestor chromosome with sufficient coverage above 20× to assign the SNP sites (646,749/657183 sites; 98.4% coverage).
Regarding coverage of the chromosome, B. anthracis Ames Ancestor and A0149 showed greater than 99% coverage, whereas B. cereus G9241 and VD166 showed less than 85% coverage, which is consistent with the fact that B. anthracis Ames Ancestor is used as a reference genome sequence. This phylogenetic distance between B. anthracis and B. cereus can be observed in Figure 1. Despite the lower coverage of B. cereus strains, 646,749 out of the 657,183 (98.4%) SNP sites were able to be assigned for construction of the phylogenetic tree (Figure 2), suggesting that the selected SNP sites were sufficient and effective for classification of B. anthracis and B. cereus species. A0149 is one of the Trans-Eurasian (TEA) group strains of B. anthracis, and the obtained phylogenetic tree (Figure 3) suggests that MiSeq raw short reads of A0149 could be correctly assigned as closely related to the Ba4599 strain from the outbreak in 2009 originating from a Scottish heroin user, as well as the report by Price et al. 11
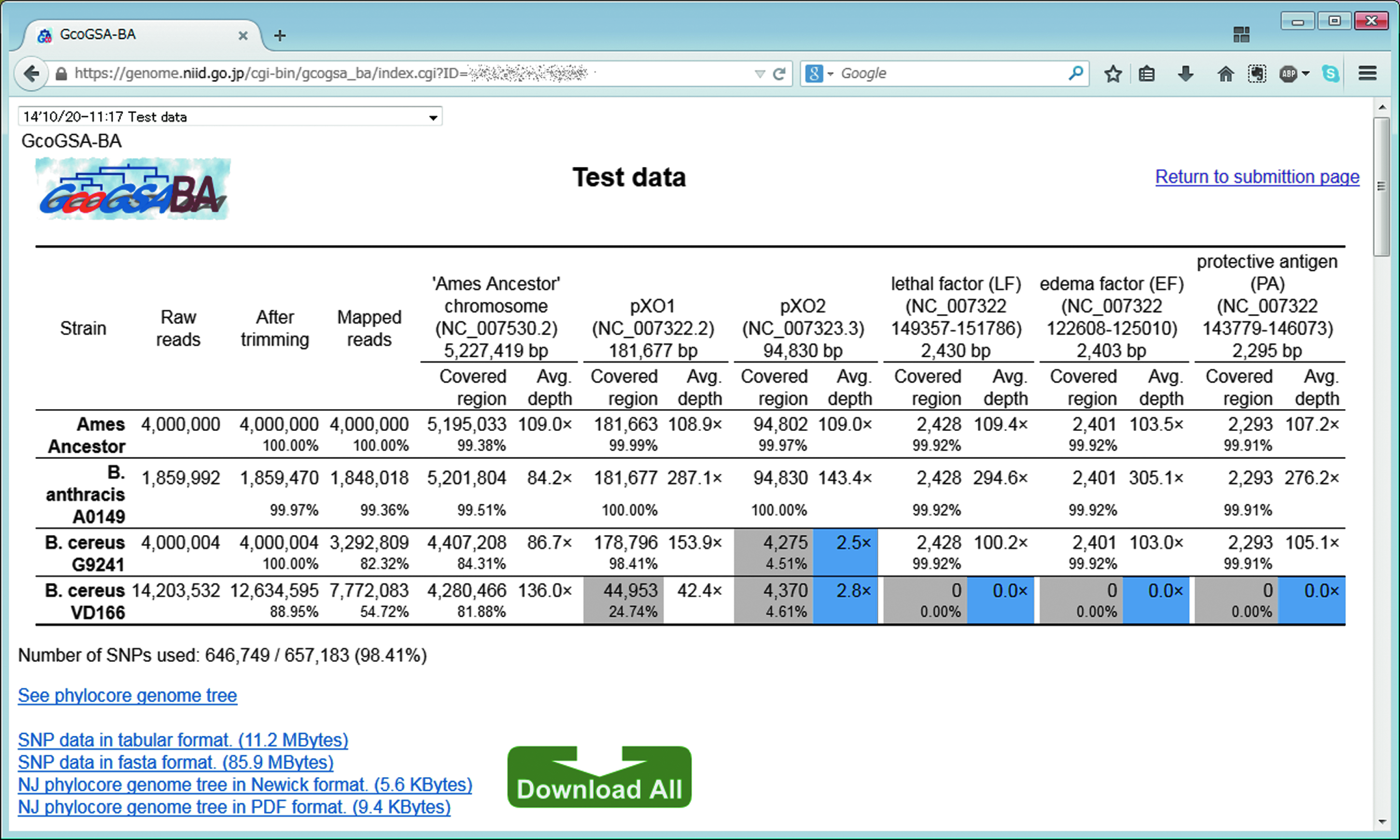
Screenshot of the results page on the GcoGSA-BA web tool. Less than 80% region coverage for the respective target sequences is shown in gray, and less than 5×read depth is shown in dark blue. All results can be retrieved from the “Download All” icon. (Color graphics available online at www.liebertonline.com/hs)
NJ phylogenetic tree generated by GcoGSA-BA. The 4 challenged data sets are shown in red. Branches corresponding to B. anthracis strains are shown in red. (Color graphics available online at www.liebertonline.com/hs)
The phylocore-genome analysis for B. anthracis strains only. (Color graphics available online at www.liebertonline.com/hs)
GcoGSA-BA could also detect both pXO1 and pXO2 sequences and 3 major anthrax toxins (LF, EF, and PA) with bwa-sw mapping. In addition to the anthrax toxin-positive Ames Ancestor and A0149 strains, the B. cereus G9241 strain showed positivity for the 3 toxins evaluated. B. cereus G9241 was isolated from the sputum and blood of a patient with life-threatening pneumonia, carrying a plasmid that is fully homologous to the pXO1 plasmid. 12 Thus, toxin-positive Bacillus strains can be identified simply using NGS and following the GcoGSA-BA web tool.
Although the NJ phylogenetic method is applied with GcoGSA-BA to facilitate rapid construction of phylocore-genome trees, the maximum likelihood method may better estimate the correct phylogenetic lineage in regard to the molecular clock. The user can further analyze the results with sufficient bootstrapping analysis on another website, called RAxML BlackBox (http://embnet.vital-it.ch/raxml-bb/), 13 using concatenated SNP alleles (“SNPs data in fasta format”) from the download site.
Footnotes
Acknowledgments
This work was supported by a grant-in-aid from the Ministry of Health, Labour, and Welfare, Japan (H23 Shinko-Ippan-007 and H26-Shinkogyosei-Shitei-002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. An icon in the web content was provided free of charge from the homepage of the materials' provider “Airuumu” (this web page is now closed). The authors declare no conflict of interest associated with this manuscript.