
Abstract
As the ability to engineer biological systems improves with increasingly advanced technology, the risk of accidental or intentional release of a dangerous genetically modified organism becomes greater. It is important that authorities can carry out attribution for the source of a genetically modified biological agent release. In the absence of evidence that ties a release directly to the individuals responsible, attribution can be carried out in part by discovering the in silico tools used to design the engineered genetic components, which can leave a signature in the DNA of the organism. Previous attribution methods have focused on identifying the laboratory of origin of an engineered organism using machine learning on plasmid signatures. The next logical step is to address attribution using signatures from the tools that are used to create the engineered modifications. A random forest classifier was developed that discriminates between design tools used to optimize coding regions for incorporation into the genome of another organism. To this end, tens of thousands of genes were optimized with 4 different codon optimization methods and relevant features from these sequences were generated for a machine learning classifier. This method achieves more than 97% accuracy in predicting which tools were used to design codon optimized genes for expression in other organisms. The methods presented here lay the groundwork for the creation of effective organism engineering attribution techniques. Such methods can act both as deterrents for future attempts at creating dangerous organisms as well as tools for forensic science.
Introduction
Technologies enabling genome engineering such as gene synthesis, clustered regularly interspaced short palindromic repeats (CRISPR), and metabolic engineering allow for changes to DNA that can create new function or alter the behavior of an organism. 1 Many of the biological systems resulting from these methods can be used for important processes contributing to the bioeconomy and betterment of the environment and society, such as bioproduction for high-value chemicals, remediation of contaminated areas, or human health.2-4 However, organism engineering can also result in the intentional modification and deployment of dangerous pathogens or ecologically damaging pests. The same methods used to develop engineered organisms intended for safe use can be employed to create altered, dangerous organisms.
As the potential for release of harmful organisms rises with the increased availability and ease of use of genome engineering techniques, 5 the tools to detect, identify, and attribute the source of these organisms will be of great importance. Such tools can act as a deterrent to, or allow for prosecution of, those responsible for the creation of dangerous engineered organisms. Before attribution can occur, detection of a dangerous organism must be carried out using genetic sequence, protein recognition, or system impact 6 in order to respond appropriately. Having detected a potential threat, identification and classification are the next steps in the analysis. Next-generation sequencing combined with bioinformatic analysis can identify known organisms and may detect engineered regions, 7 although such synthetic DNA components are not often considered in traditional bioinformatic processes. Methods using machine learning to determine whether a suspected organism is engineered have been created, 8 but use of such technologies by forensic science will likely benefit from further development.
Once the determination has been made that an organism is dangerous and has been engineered, it is important that scientists are able to identify who is responsible, and uncovering where or how such an organism was created can aid in that process. Previous efforts on engineering attribution have focused on the laboratory of origin of plasmids.9-13 Using DNA sequence data from the Addgene plasmid repository and associated metadata, including the laboratory that developed the plasmid, classifiers were able to accurately identify the laboratory of origin, country of origin, and laboratory lineage. These existing methods rely on data from engineered plasmids, which are not always ideal for use in organism engineering. In particular, plasmids can be unstable if they create a metabolic burden on the organism in which they reside, and the copy number of this DNA can be difficult to control.14,15 Plasmids were traditionally considered less complicated to engineer than genomes, 16 but with the advent of genome engineering techniques such as DNA synthesis, advanced assembly methods, and CRISPR, the barrier to genome editing has lowered. Genome engineering-focused and agent agnostic attribution capabilities are necessary because an organism engineer may not use plasmids or published sequences. The creation of such methods will add to the capability to identify the designer of the engineered organism (but not necessarily those who carry out an attack with such an agent).
The majority of engineering biology occurs at the DNA level and utilizes the general engineering framework of the design, build, test, and learn cycle. The first 2 phases of this framework use methods that can leave signatures in modified genetic material. For instance, gene synthesis, assembly, or genome engineering 17 can leave traces in the nucleotide sequence based on the processes used to print, cut, and stitch together the DNA. Similarly, the tools used to design DNA to be altered in a genome18,19 can leave signatures due to the design choices and necessary elements for incorporation into the organism. For example, gene sequences for heterologous expression are often “codon optimized” for efficient expression, or to facilitate DNA synthesis and assembly, among other goals. 20 This optimization can be designed in silico at the codon level by swapping synonymous codons for the existing sequence, usually based on the organism in which the genes will be expressed. These methods for codon optimization can use different algorithms and therefore would produce unique signatures that could be categorized by the design tool used.
With the tools that are used for engineering biology described above, the possible design space that can be explored is vast. In attempting to carry out attribution for a biological attack, the large sequence and functional space that can be leveraged by biological engineers, as well as a paucity of information on the tendencies and methods used by nefarious actors, can complicate the process. For these reasons, there is a need for machine learning methods that can identify the tools used for organism engineering. To address these needs, this study employed a machine learning classifier to identify codon optimization methods used for heterologous gene expression in engineered organisms.
Materials and Methods
Data Collection
All coding sequences (CDSs) from the genomes of multiple organisms were downloaded from GenBank including those from Escherichia coli MG12655, 21 Bacillus subtilis PS216, 22 Saccharomyces cerevisiae S288C, 23 Caenorhabditis elegans WBcel235, 24 Drosophila melanogaster, 25 Mus musculus strain C57BL/6J, 26 and Homo sapiens GRCh38.p13. 27 The number of CDSs for each of these downloads are indicated in the Table.
Number of Coding Sequences Collected From Organisms Used in This Study
Optimization
Code for 4 different optimization algorithms was downloaded and installed on the Massachusetts Institute of Technology Lincoln Laboratory supercomputing grid. 28 The codon optimization algorithms used were DNA Chisel 29 from the Edinburgh Genome Foundry at the University of Edinburgh, Optipyzer, 30 Simple Codon Optimizer, 31 and Codon Harmonizer. 32 All codon optimization tools used rely on the same few algorithmic principles. Codon Harmonizer and Optipyzer allow for codon harmonization where the tool finds a balance between the source and destination organism codon usage. DNA Chisel and Simple Codon Optimizer carry out codon optimization where the algorithm only optimizes for the destination organism codon usage. This is discussed in more detail in the Supplemental Description (www.liebertpub.com/doi/suppl/10.1089/hs.2022.0152).
Each of the CDSs from E coli, B subtilis, and S cerevisiae were optimized for expression in 7 different organisms (E coli, B subtilis, S cerevisiae, C elegans, D melanogaster, M musculus, H sapiens) including optimization for the organism from which the sequences originated. The optimization algorithms were run in as simple a manner as possible, including avoiding extra parameters that may be allowed by the algorithm but that might skew the results and create an easier classification problem. The parameters with which the algorithms were run are shown in the supplemental description. In some cases, the codon tables were hard coded into the method (as in the DNA Chisel and Optipyzer algorithms) and in others the codon table is provided by the user (Simple Codon Optimizer) or calculated from the original sequence set for that organism (Codon Harmonizer). In the user-provided case, the codon table most similar to those used in hard-coded cases was chosen.
Feature Generation
A number of features were generated for each of the optimized gene sequences. The features chosen represent aspects of genetic sequence commonly used in codon optimization methods 33 that could be reasonably calculated for a wide variety of coding regions for many organisms. All data were generated in Python version 3.8 (Python Software Foundation, Beaverton, OR) using codon optimized sequences as input. G and C nucleotide (GC) content was calculated using the GC function of the BioPython Bio.SeqUtils package, while codon adaptation index 34 (CAI) was generated with the Bio.SeqUtils.CodonUsage class. For this study, the same reference codon table was used to calculate CAI for all samples, and assumed equal use of each codon. Codon usage (representation of each codon in a sequence by percentage) was calculated by tallying the occurrence of each codon in a list, and dividing by one-third of the length of the full sequence. The feature for the longest homopolymer finds the longest string of a repeated nucleotide and returns the identity of that nucleotide and the length of the repeated bases. Similarity to the wild-type sequences was calculated using the Bio.pairwise2.align.globalxx method. Additional features were generated by byte pair encoding (BPE) as discussed in the following section on machine learning. All features used for training the random forest algorithm are shown in the first column of Supplemental Table S1 (www.liebertpub.com/doi/suppl/10.1089/hs.2022.0152).
Machine Learning
BPE 35 was carried out using the Python implementation of SentencePiece 36 to find DNA motifs that may function as features contributing to the classification of optimization methods. The wild-type sequences of all CDSs from all organisms in the Table were used as a training set to generate the motifs. The vocabulary size of this analysis was set to 1,000 motifs based on previous methods in which BPE was employed to discover DNA motifs. 10 The resulting vocabulary consists of DNA sequences from 1 to 9 base pairs. Each of these short DNA sequences was used as a feature for the classifier where the number of occurrences of each of the motifs was counted for all optimized genes individually. These features are sparsely populated as many motifs do not appear in a given sequence.
Classification was carried out with the random forest implementation in Python (RandomForestClassifier) from the scikit-learn library. 37 The complete dataset consisted of 411,796 samples, each with 1,069 features. The training and test set data split was 80% training data and 20% test data (329,436 and 82,360 samples, respectively) using scikit-learn's train-test-split module. The number of sequences from each organism in the training and test data are in Supplemental Table S2 (see Supplemental Tables S2 to S5 at www.liebertpub.com/doi/suppl/10.1089/hs.2022.0152). Parameter optimization for number of trees (n estimators), maximum allowed depth of the tree (max depth), and the maximum number of features considered for random sampling at each branch point (max features) was performed by training the classifier, on training data only, using a range of values for the above parameters and reporting out-of-bag (OOB) error. The OOB error is an informative metric of a random forest model that allows for the classifier to be fit and validated during training. The parameters that yielded the lowest OOB error were chosen for subsequent use: 400 trees, a maximum depth of 5, and number of features limited to the square root of the total number of features. The random forest classification was run with the Gini impurity measure. All classifier runs were performed on the Lincoln Laboratory supercomputing grid with 4 Intel Xeon Platinum 8,260 CPUs and 4 GB of RAM per core using Python version 3.8.
Feature selection was performed after the initial training of the model exclusively on the training data. Recursive feature elimination (RFE) was used to reduce the feature set. This process included first training a random forest classifier with all features from the training data. Next, features were ranked by their importance, and 20% of the features were removed from the training data based on lowest importance. Then, the classifier was retrained and the new feature importances were calculated. This process was repeated until the OOB error reached an approximate lower bound and the features used at that point were determined to be the optimal reduced feature set. Additive feature determination was also carried out where feature importance scores were reported from the random forest model and features were ranked by order of importance. Next, a new random forest was trained on a subset of the most important features (adding 5 features at a time by order of importance and then 1 feature when approaching the highest accuracy) and OOB score was reported at each iteration.
Results
The overall method to create this classifier is shown in Figure 1. The process flow starts with gathering sequences for many genes from different organisms. The DNA sequences are then optimized by 4 different algorithms for expression in other organisms including the organism from which the genes originated. Features are generated from the optimized sequences such as codon usage, GC content, and DNA motifs determined by machine learning, among others. The features used were those sequence attributes that codon adaptation is used to address (eg, GC content, codon usage, homopolymers) or those that result from the codon optimization (eg, CAI, codon usage, similarity to wild-type). These feature data are separated into training and test sets. Using these feature sets, a random forest classifier is created from the training set.
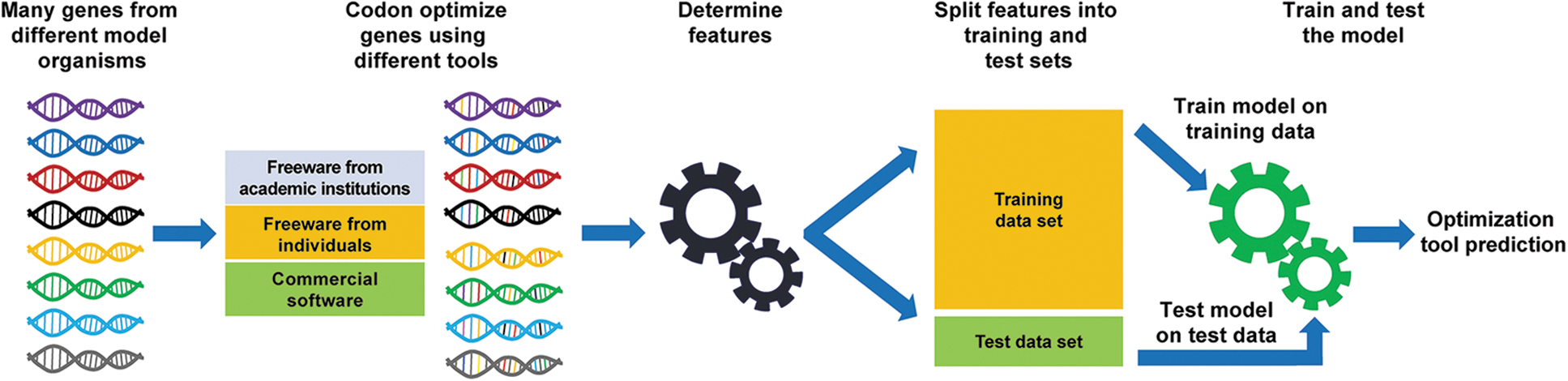
Process flow for collecting data, generating features, determining features, training and testing the classifier, as well as predicting the optimization tool used for each sample.
Hyperparameter Tuning
To construct the most effective classifier, hyperparameter tuning can be used to reduce classification error. Here, the n estimators and max features were tuned to reduce the classifier error rate as discussed in the methods section. This parameter tuning is shown in Figure 2. The effect of the n estimators and the max features on OOB error rate is considered in Figure 2A. Using the square root of the total number of features as the max features parameter allows the classifier to achieve a lower OOB error rate of those tested. The number of estimators to use for the classifier was determined to be 400, as there is little to no decrease in OOB error rate after this value. The value of the max depth was set at 5 as little decrease in OOB error is indicated after this point (Figure 2B).

Hyperparameter tuning of max features, number of estimators, and tree depth using OOB error. (A) Number of estimators vs OOB error rate for the square root of the total features (blue) and log2 of the total features (red). (B) Tree depth vs OOB error rate for the square root of the total features (blue), log2 of the total features (red), and the total features (green). Abbreviations: OOB, out of bag; sqrt, square root.
Feature Selection
To discover a reduced set of features that reach a similar accuracy to the total set and avoid overfitting, features were ranked by their importance as calculated by the random forest classifier (Supplemental Table S1). Classifiers were then trained on subsets of the most important features. The results of the RFE are shown in Supplemental Figure S1 (see all Supplemental Figures at www.liebertpub.com/doi/suppl/10.1089/hs.2022.0152) indicating that the OOB error levels off to a lower bound between 40 and 50 features. Similarly, the additive feature determination process is shown in Figure 3A. Comparison of the number of ranked features and the OOB score showed that the OOB score reaches 96% at 45 features and plateaus at an OOB score of 97.52% with the full feature set. The top 45 feature minimal classifier includes BPE motifs, codon usage features, the GC content, and the CAI. Figure 3B shows the top 20 features ranked by importance and the distribution of those features across the different classes. This distribution represents the mean of the codon frequencies (number of appearances of a particular codon in a gene divided by the gene length) for all genes optimized by one of the codon optimization algorithms. The tenth most important feature (CGGT) is shown separately as the scale of this feature was significantly higher than the others. CGGT is a feature discovered with BPE and is therefore a count of appearances of that motif in each sequence, whereas the codon usage features that make up the rest of the top 20 features are all less than or equal to 1.

Feature selection for the random forest classifier. (A) The number of features vs OOB score. (B) Feature distribution across the 4 different algorithms for the top 20 features by importance. The BPE feature CGGT pattern is separate due to the scale skewing the visual result of the other features. Abbreviations: BPE, byte pair encoding; OOB, out of bag.
Classifier Performance
The random forest model trained on the full dataset (1,069 features) reached an overall test accuracy of 97.76%. Similarly, the OOB score was 97.52%. The confusion matrix of the results indicates strong ability to distinguish between the 4 classes (Figure 4). Error for predictions was very low across all classes. DNA Chisel and Simple Codon Optimizer were most often misclassified as one another (4.4% of the samples). DNA Chisel was misclassified as simple-codon-optimizer in 2.9% of predictions. All other incorrect classifications occurred less than 1% of the time. Supplemental Table S3 shows the precision and recall for the 4 classes in the classifier.

Confusion matrix for the random forest classifier using all features. The numbers in each box represent the proportion of the classifications where the true label was classified as the predicted label. Abbreviations: BPE, byte pair encoding; OOB, out of bag.
The classifier was also tested on E coli, B subtilis, and S cerevisiae genes optimized for expression in G gallus, an organism for which the classifier was not trained (Supplemental Table S4 and Supplemental Figure S2). There is a slight drop in performance to an accuracy of 94.01% and OOB score of 95.48% but the classifier still performs quite well.
Discussion
This work represents a demonstration of the capability of machine learning to identify in silico tools that were used to design synthetic DNA for organism engineering. Knowledge of the tools used in gene design could be leveraged by forensic scientists to aid in attribution in the event of the release of a dangerous pathogen or environmentally harmful organism. 38 For instance, a recorded download of an in silico tool by a potential perpetrator could be associated with the released organism that was designed with that tool. Similarly, synthesis providers often have unique design algorithms. DNA synthesis orders by a suspected actor could be connected to a particular biological agent if the organism has the DNA signatures from the synthesis provider's design method. These connections could potentially contribute to evidence for attribution.
The optimization methods used in this study were those that are well supported and can handle high-throughput optimization of coding sequences. Many codon optimization software tools exist, but few met all the requirements for this work. In order to generate the large synthetic dataset needed for training our model, a tool that could function in a high-throughput manner (or was open source and could be modified to do so) was required. This eliminated all tools offered through companies (mostly DNA synthesis companies), such as Integrated DNA Technologies, GenScript, and Twist Bioscience. While popular, these would not have been able to process thousands of sequences in high-throughput, and their source code is proprietary and not open source. Other optimization tools with a lack of support often resulted in difficulty installing and running programs. The 4 tools selected represent a diversity of codon optimization algorithm type (harmonization and optimization), authorship (large research institution vs a single researcher), and popularity (widely distributed in the Benchling Molecular Biology Suite software or limited adoption on GitHub, Supplemental Table S5). The availability and support limitations of many of the optimization tools may mean that organism engineers would be inclined to choose from the limited set of the most useful available tools, simplifying the attribution of design methods. It is also possible that an engineer might choose a rarely used optimization tool or create their own. While this would pose a unique challenge, machine learning might still be capable of determining the type of algorithm used for optimization (eg, a particular type of codon harmonization) or discover unique signatures of a truly novel method.
The use of a random forest classifier offered advantages over other machine learning methods. Deep learning has been used for classification of this type, 9 which can have the benefit of discovering features that might not otherwise be used. However, it can be difficult to deduce which aspects of the data have contributed to the classification. The choice of the supervised random forest classifier method was due in part to its ability to give insight into how each feature contributes to the classification of the samples through feature importance scores. Such insight can be impactful in forensic science attribution where decision criteria can be of legal and ethical importance. 39 Supplemental Table S1 indicates that the relevant features, ranked by importance, are a combination of codon usage, motifs discovered by BPE, and calculated features such as CAI and homology to wild-type. Discovering which of these features are important without machine learning would be a labor-intensive process that may not result in the most accurate classifier. Feature generation, rather than using the sequence itself, also serves to avoid data leakage between training and test sets, as the genomes of the source organisms chosen likely have some level of similarity across genes.
In an additional attempt to avoid overfitting, we sought to eliminate extraneous features, which is a concern when features may be correlated. 40 To this end, we used the approach of RFE, 41 as well as additive feature determination. Upon evaluation of accuracy for each model, we determined that using 45 features maximized accuracy while minimizing the number of features (Figure 3A). The balance between minimization of features and the level of accuracy that is reached is dependent on the application. Here, there is very little impact to the accuracy with features that are limited to 4% of the original feature set, decreasing the chance of highly correlated features.
When gauging the usefulness and performance of a classifier, it is also important to determine if choices made by the user during training and testing have impacted the results. One factor that can impact the classification of the codon optimization algorithms are the codon usage tables. If all of the codon tables used by optimization algorithms are drastically different, this single factor can artificially simplify the classification problem during training and testing. As an example, the use of different codon usage tables as part of their algorithms results in the misclassification of Codon Harmonizer as DNA Chisel only 0.5% of the time, while DNA Chisel is never incorrectly classified as Codon Harmonizer (Figure 4). However, the classifier is still capable of correctly classifying the optimization tool used even when the codon usage tables are the same. DNA Chisel was misclassified as the Simple Codon Optimizer only 2.9% of the time, while the Simple Codon Optimizer was classified as DNA Chisel only 4.4% of the time using the same codon usage tables. This indicates that algorithmic differences other than the codon table, such as codon harmonization (Codon Harmonizer) versus optimization (DNA Chisel), are also important to the classification. The ability to differentiate between classes without having to rely on a user choice, such as the codon table used, results in a method that is more robust and generally applicable.
The ability to classify a set of optimization algorithms with high accuracy, as demonstrated in this methodology, could be extended to other aspects of organism genome engineering to further assist attribution. Beyond in silico tools, it may be possible to extend this methodology to estimate the experimental methods used to engineer a cell. For example, certain nucleases or homologous recombination methods may leave signatures that can help in determining how an engineered organism was created. Data collection on design and construction of engineered organisms and the resulting DNA signatures will enable future attribution methods.
Conclusion
While recent biotechnological advancements are largely directed toward beneficial outcomes, they may also facilitate the engineering of dangerous organisms. The development of computational tools to determine how an engineered organism was created can provide information to aid attribution and potentially act to deter nefarious actors. Machine learning is well suited for the task of discriminating between different methods used to design and build engineered organisms. In this work, a random forest classifier was able to correctly identify, with high accuracy, the method of codon optimization (of 4 total) used to design a sequence of DNA for heterologous expression in an engineered organism. Hyperparameter tuning and feature minimization for the classifier helped decrease error and avoid overfitting from highly correlated features, respectively. The result is a classifier capable of discriminating between the codon optimization methods with an accuracy of over 97%. This demonstrated that machine learning can play and important role in providing information for attribution of genetically engineered organisms, and serves as an example for further development of such computational tools.
Footnotes
Acknowledgments
The authors would like to acknowledge Josh Dettman for help in developing the concept and overseeing administrative requirements, and Chelsea Lennartz and Rafael Jaimes for editing the manuscript. This material is based upon work supported by the Defense Threat Reduction Agency and the Under Secretary of Defense for Research and Engineering under Air Force Contract No. FA8702-15-D-0001. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the Defense Threat Reduction Agency. DISTRIBUTION STATEMENT A. Approved for public release. Distribution is unlimited.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.