
Abstract
Adeno-associated virus (AAV)-mediated microdystrophin gene therapy holds great promise for treating Duchenne muscular dystrophy (DMD). Previous studies have revealed excellent skeletal muscle protection. Cardiac muscle is also compromised in DMD patients. Here we show that a single intravenous injection of AAV serotype-9 (AAV-9) microdystrophin vector efficiently transduced the entire heart in neonatal mdx mice, a dystrophin-deficient mouse DMD model. Furthermore, microdystrophin therapy normalized the heart rate, PR interval, and QT interval. The cardiomyopathy index was also significantly improved in treated mdx mice. Our study demonstrates for the first time that AAV microdystrophin gene therapy can ameliorate the electrocardiographic abnormalities in a mouse model for DMD.
Introduction
Dystrophin is a 427-kDa subsarcolemmal protein. It links the extracellular matrix with cytoskeletal F-actin in striated muscle. A major biological function of dystrophin is to dissipate contraction-associated mechanical stress and stabilize the sarcolemma. This is achieved through the assembly of the dystrophin-associated glycoprotein complex (DGC) at the sarcolemma. In particular, the dystrophin N-terminal domain and a region of the rod domain independently bind to cytosolic F-actin and the dystrophin cysteine-rich domain binds to the transmembrane dystroglycan complex. These interactions link the cytoskeleton with the extracellular matrix (Blake et al., 2002).
The large size of the full-length dystrophin coding sequence has been a longstanding challenge for DMD gene therapy (Duan, 2006b). It exceeds the packaging capacity of many viral vectors. To overcome this obstacle, investigators have developed highly truncated but partially functional minigenes and microgenes (Harper et al., 2002). The ΔR4-23/ΔC microgene (ΔR4/ΔC) is one of the best characterized microgenes. Local and systemic delivery of the ΔR4/ΔC microgene with recombinant adeno-associated viral vector (AAV) has resulted in significant improvement in skeletal muscle pathology and muscle force (Harper et al., 2002; Gregorevic et al., 2004, 2006; Liu et al., 2005; Yue et al., 2006).
Two studies evaluated the ΔR4/ΔC microgene in the heart of mdx mice, a dystrophin-null mouse model for DMD. Direct cardiac injection of AAV serotype-5 (AAV-5) microgene vector strengthened cardiomyocyte sarcolemmal integrity in neonatal mdx mice (Yue et al., 2003). Limited hemodynamic improvement was also documented when AAV serotype-6 (AAV-6) microgene vector, vascular endothelial growth factor (VEGF), and heparan sulfate were codelivered to mdx mice (Townsend et al., 2007). These studies also showed that microdystrophin can stabilize the sarcolemmal DGC in the mdx heart (Yue et al., 2003; Townsend et al., 2007).
Among different AAV serotypes, AAV serotype-9 (AAV-9) is by far the most potent serotype for myocardial transduction when tested in normal mice (Inagaki et al., 2006; Pacak et al., 2006; Bostick et al., 2007; Ghosh et al., 2007). However, it is not clear whether AAV-9 can efficiently transduce the myocardium in diseased mice. Here we tested cardiac AAV-9 transduction in neonatal mdx mice. These results suggest that a single intravenous injection in the absence of additional pharmacological agents can lead to robust microdystrophin expression throughout the entire heart of newborn mdx mice. To determine whether early intervention can lead to functional improvement, we examined the electrocardiogram (ECG) profile in AAV-9-treated mice. Unlike symptomatic human patients, mdx mice do not display severe heart disease until they are old (∼2 years of age) (Bostick et al., 2008). However, young mdx mice do display the characteristic ECG changes seen in preclinical-stage human DMD patients. These changes include a shortened PR interval, a prolonged QT interval, and tachycardia (Heymsfield et al., 1978; Nigro et al., 1990; Chu et al., 2002; Sadeghi et al., 2002; Markham et al., 2005). Four months after AAV-9 therapy, heart rate and PR and QT intervals were all normalized in treated mdx mice. These results are consistent with previous reports and together they suggest that microdystrophin gene therapy holds great promise in ameliorating both skeletal and cardiac muscle diseases in DMD.
Materials and Methods
Animals
All animal experiments were approved by the Animal Care and Use Committee of the University of Missouri (Columbia, MO) and were in accordance with the National Institutes of Health (NIH, Bethesda, MD) guidelines. C57BL/10SnJ (BL10) and dystrophin-deficient C57BL/10ScSn-Dmdmdx /J (mdx) mice were originally purchased from Jackson Laboratory (Bar Harbor, ME). Experimental mice were obtained from local breeding colonies. All mice were housed in specific-pathogen free animal care facilities at the University of Missouri and kept under a 12 hr light (25 lux):12 hr dark cycle with free access to food and water.
Recombinant AAV-9 vector
AAV-9 vectors were produced according to a previously reported triple-plasmid transfection protocol (Bostick et al., 2007; Ghosh et al., 2007). Briefly, 70% confluent 293 cells were transfected with a microdystrophin cis-plasmid (pcis.CMV.ΔR4/ΔC), a pRep2/Cap9 helper plasmid (a gift from J. Wilson, University of Pennsylvania, Philadelphia, PA) (Gao et al., 2003, 2004), and an adenoviral helper plasmid (pHelper; Stratagene, La Jolla, CA) at a ratio of 1:3:3. The ΔR4/ΔC microgene was a gift from J. Chamberlain (University of Washington, Seattle, WA). This microgene is based on the human dystrophin gene and it carries only ∼30% of the full-length dystrophin-coding sequence. The ΔR4/ΔC microgene contains regions from the N-terminal domain to hinge 2 and from spectrin-like repeat 24 (R24) to the cysteinerich domain (Harper et al., 2002). Recombinant viral stocks were purified through two rounds of isopycnic CsCl ultracentrifugation as described previously (Lai et al., 2005). Viral titration and quality control were performed according to a previously published protocol (Xu et al., 2004; Lai et al., 2005).
In vivo gene delivery
AAV-9 microdystrophin vector was injected into conscious 1-day-old mdx mice in a single 100-μl bolus through the facial vein according to a previously described protocol (Ghosh et al., 2007). Each mouse received 1 × 1012 vector genome (VG) particles of AAV-9.
Immunofluorescence staining
Microdystrophin expression was determined by immunofluorescence staining at 4 months of age, using a human dystrophin-specific antibody (Dys-3, diluted 1:20; clone Dy10/12B2, IgG2a; Novocastra, Newcastle, UK). Immunostaining was performed essentially as described previously (Yue et al., 2003, 2006).
ECG evaluation of heart function
A noninvasive 12-lead ECG was performed in 4-month-old age- and sex-matched mice including BL10, mdx, and AAV microgene-treated mdx. ECG tracings were sequentially recorded from each lead and analyzed according to a previously published protocol (Bostick et al., 2008). Briefly, cardiac electric activity signals were processed through a single-channel bioamplifier (model ML132; ADInstruments, Colorado Springs, CO) and then recorded on a model MLA0112S PowerLab system using Chart software (version 5.5.5; ADInstruments). Beats from a continuous 1-min recording were averaged to generate a signal-averaged ECG for analysis with Chart ECG analysis software (version 2.0; ADInstruments). The amplitude of the Q wave was analyzed with lateral limb lead (I and aVL) and left precordial lead (V5 and V6) tracing. The remaining ECG parameters were analyzed on the basis of lead II tracing results. The heart rate-corrected QT interval (QTc) was calculated according to Mitchell and coworkers (1998). The cardiomyopathy index was determined by dividing the QT interval by the PQ segment (QT/PQ).
Statistical analysis
Data are presented as means ± standard error of mean. Statistical analysis was performed with SPSS software (SPSS, Chicago, IL) using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc analysis. Differences were considered significant when p < 0.05.
Results and Discussion
We first tested whether systemic AAV-9 delivery can efficiently transduce a dystrophin-deficient heart. The transduction level needed to correct mdx cardiomyopathy remains to be determined for the microdystrophin gene. In the case of full-length dystrophin gene, expression in 50% cardiomyocytes has been shown to normalize heart function (Yue et al., 2004; Bostick et al., 2008). Because the microgene is less competent than the full-length gene, we speculated that greater than 50% transduction would be required to achieve a therapeutic effect. Among available AAV serotypes, AAV-6 and AAV-8 are the only ones that have been shown to transduce the whole heart in dystrophic animals (Zhu et al., 2005; Gregorevic et al., 2006; Townsend et al., 2007). However, AAV-6 transduction requires coadministration of VEGF (Gregorevic et al., 2006; Townsend et al., 2007). Interestingly, side-by-side comparison studies from two groups suggest that AAV-9 is 5- to 10-fold stronger than AAV-8 in transducing the normal mouse heart (Inagaki et al., 2006; Pacak et al., 2006). The cardiac tropism of AAV-9 has since been confirmed by studies from several groups (Bostick et al., 2007; Ghosh et al., 2007; Miyagi et al., 2008; Zincarelli et al., 2008). On the basis of these observations, we hypothesized that AAV-9 should result in robust cardiac transduction in mdx mice.
Four months after a single intravenous injection of 1 × 1012 VG particles of AAV-9 microdystrophin vector, we examined microgene expression in the heart (Fig. 1). We observed efficient microdystrophin expression throughout the entire heart. The left ventricle, right ventricle, and septum were saturated with microgene-expressing cardiomyocytes (Fig. 1). These results demonstrated for the first time that AAV-9 is not only a superior vector for the normal heart, it also transduces the diseased heart at levels that should meet many therapeutic applications.

A single intravenous AAV-9 injection leads to efficient microdystrophin expression throughout the entire heart. Representative immunofluorescence staining images of the mdx heart infected with AAV-9 microdystrophin vector (n = 12). mdx mice were infected at 1 day of age and immunofluorescence staining was performed 4 months after AAV infection with a human dystrophin-specific antibody. High-magnification photomicrographs for the left ventricle (LV), right ventricle (RV), and septum (Sep) were obtained from the respective boxed areas in the full-view image.
Next, we examined whether neonatal microdystrophin therapy could ameliorate electrophysiological abnormalities of the mdx heart. Despite the lack of dystrophin, young adult mdx heart exhibits minimal histopathology (Bridges, 1986; Coulton et al., 1988; Grady et al., 1997; Kamogawa et al., 2001; Nakamura et al., 2002; Hainsey et al., 2003). Consistent with these reports, we also did not see apparent fibrosis and inflammation in 4-month-old mdx hearts (data not shown). Studies from us as well as other investigators suggest that young mdx mice have a normal hemodynamic profile unless challenged with positive inotropic drugs such as dobutamine and β-isoproterenol (Quinlan et al., 2004; Yue et al., 2004). ECG abnormalities are the only consistent cardiac findings in young mdx mice. The heart rate of young mdx mice is significantly higher than that of normal BL10 mice (Chu et al., 2002). Conduction through the atrioventricular (AV) node is also accelerated, as reflected by a shortening of the PR interval. (Note: Other types of conduction defects such as bundle branch block, AV block, sinoatrial [SA] block, and ventricular arrhythmias have also been reported in symptomatic DMD patients and old mdx mice [Wehling-Henricks et al., 2005].) Furthermore, the QT interval, which represents the duration of ventricular depolarization and repolarization, is prolonged in young mdx mice (Sadeghi et al., 2002). Importantly, the ECG changes in young mdx mice mirror the findings seen in DMD patients before they develop symptomatic heart disease (Heymsfield et al., 1978; Nigro et al., 1990; Steare et al., 1992; Oguz et al., 1998; Markham et al., 2005).
Consistent with previous reports, we observed tachycardia, PR interval reduction, and QTc (heart rate-corrected QT interval) prolongation in young mdx mice (Fig. 2). Unlike old mdx mice, the QRS duration (lead II) and the Q wave (leads I, aVL, V5, and V6) were not altered in young mdx mice (Fig. 2) (data not shown for Q waves from leads aVL, V5, and V6) (Bostick et al., 2008). AAV-9 microdystrophin therapy completely normalized the heart rate, PR interval, and QTc interval. There were no statistically significant differences between the treated mdx mice and normal BL10 mice in these parameters (Fig. 2C).
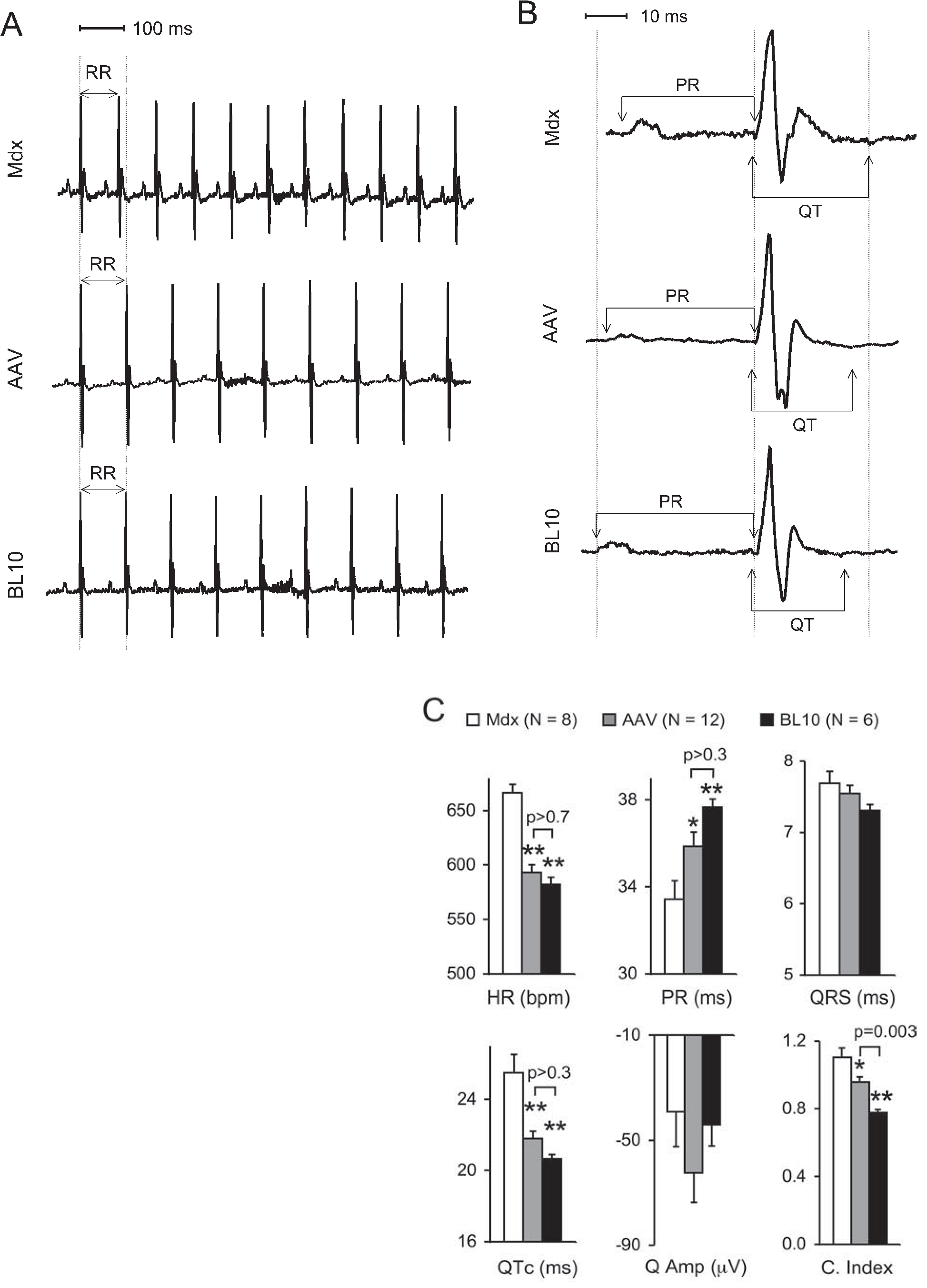
Neonatal AAV-9 microdystrophin therapy improves the ECG profile in young adult mdx mice. (
The cardiomyopathy index is defined by the ratio of the QT interval over the PQ segment. This index has been used frequently in clinical studies to identify early and/or sub-clinical cardiac involvement in DMD patients (Nigro et al., 1990; Steare et al., 1992; Oguz et al., 1998). We have shown an increase of the cardiomyopathy index in old mdx mice (Bostick et al., 2008). Similar to what has been reported in humans, we found that young mdx mice also exhibited a significantly higher cardiomyopathy index (Fig. 2C). After AAV microgene therapy, the cardiomyopathy index was significantly reduced compared with that of mdx mice. Surprisingly, however, the cardiomyopathy index was not completely normalized in AAV-treated mdx mice. This may reflect a limitation of the highly truncated microgene. Taken together, our results suggest that the microdystrophin may ameliorate but not cure Duchenne cardiomyopathy even if delivered at the neonatal stage.
Ameliorating cardiomyopathy is an important goal for DMD gene therapy. Here we have provided the first evidence that microdystrophin therapy can improve the ECG profile of mdx mice before they develop clinically evident cardiomyopathy. The pathophysiology underlying abnormal ECG findings in preclinical DMD patients and in young mdx mice remains to be characterized. Interestingly, the ECG profile mimicks what has been reported in the Lown–Ganong–Levine (LGL) syndrome, a class of preexcitation syndromes (Lown et al., 1952). No structural abnormalities have been identified in the LGL syndrome. In young mdx hearts, we also did not see prominent histopathology (data not shown). It is possible that subtle changes in dystrophin-deficient cardiomyocytes (such as channel activities, etc.), rather than myocardial fibrosis and inflammation, may have altered normal electrical signal spreading through the cardiac conduction system in young mdx mice.
In summary, we have demonstrated efficient AAV-9 transduction in the dystrophin-deficient heart. Our results also suggest that microdystrophin therapy can significantly improve electrophysiological defects in mdx mice. Because of its noninvasive nature and convenience, the ECG protocol described here will be a useful tool for monitoring cardiac function when exploring other therapeutic interventions.
Footnotes
Acknowledgments
The authors thank Dr. James Wilson (University of Pennsylvania) for providing AAV-9 packaging plasmid. The authors also thank Dr. Jeffrey Chamberlain (University of Washington) for providing the ΔR4/ΔC microdystrophin gene. This work was supported by grants from the National Institutes of Health (AR-49419; NS-62939; D.D.) and the Muscular Dystrophy Association (D.D.). B.B. was partially supported by a training grant from the National Institutes of Health (GM 008396).