
Abstract
Although the desire to develop gene therapy for hemophilia B is high, safety remains a concern. Therefore, improving the therapeutic index of gene therapy vectors is an important goal. Thus, we evaluated the use of three bioengineered factor IX (FIX) variants with improved catalytic activity in the context of the helper-dependent adenoviral vector. The first vector expressed R338A-FIX, an FIX variant with the arginine at position 338 changed to an alanine, which resulted in a 2.9-fold higher specific activity (IU/mg) compared with the wild-type FIX. The second vector expressed FIXVIIEGF1, a variant with the EGF-1 domain replaced with the EGF-1 domain from FVII, which resulted in a 3.4-fold increase in specific activity. The third expressed R338A + FIXVIIEGF1, a novel variant containing both aforementioned modifications, which resulted in a 12.6-fold increase in specific activity. High-level, long-term, and stable expression of these three variants was observed in hemophilia B mice with no evidence of increased thrombogenicity compared with wild-type FIX. Thus, these bioengineered FIX variants can increase the therapeutic index of gene therapy vectors by permitting administration of lower doses to achieve the same therapeutic outcome. Furthermore, these variants may also be valuable for recombinant FIX protein replacement therapy.
Introduction
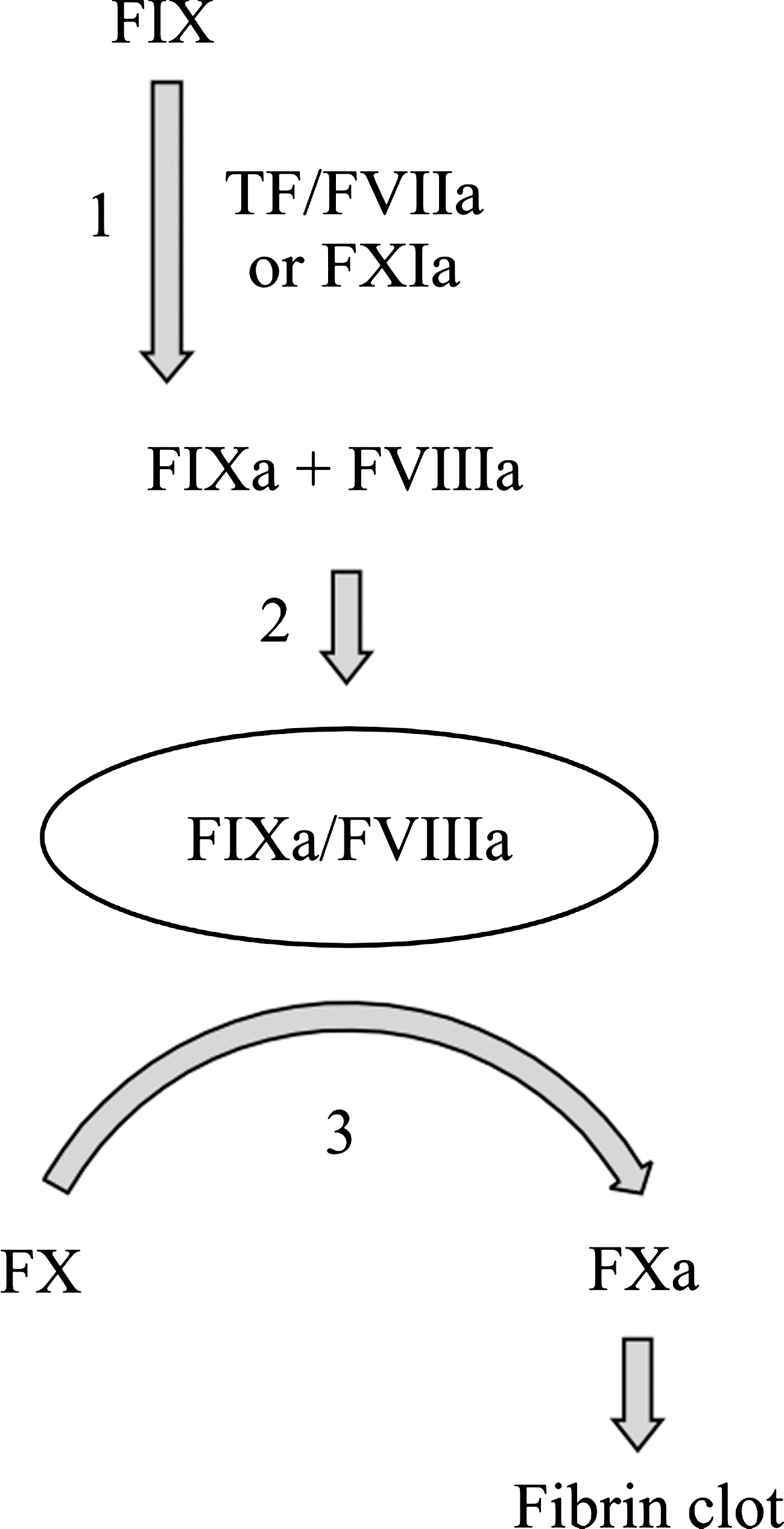
Simplified diagram showing the role of coagulation factor IX (FIX) in the clotting cascade. FIX is activated to FIXa by FXIa or tissue factor–FVIIa complex (step 1). FIXa forms a complex with FVIIIa (step 2) to activate FX to FXa (step 3), which ultimately leads to the formation of a fibrin clot. R338A-FIX enhances step 3 and FIXVIIEGF1 enhances step 2. The novel double mutant R338A + FIXVIIEGF1 is hypothesized to enhance both steps 2 and 3. TF, tissue factor.

HDAd containing wild-type (wt) FIX, R338A-FIX, FIXVIIEGF1, or R338A + FIXVIIEGF1 expression cassettes. All expression cassettes contain the liver-specific phosphoenolpyruvate carboxykinase (PEPCK) promoter, the apolipoprotein AI (ApoAI) intron, the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), the ApoE locus control region (LCR), and the human growth hormone polyadenylation region [p(A)]. The adenoviral inverted terminal repeats (ITRs) and packaging signal (ψ) are shown. Not drawn to scale.
Because the mechanisms of enhancement of R338A and FIXVIIEGF1 do not overlap (Fig. 1), we hypothesized that a novel double mutant combining both the R338A-FIX and FIXVIIEGF1 mutations will have even greater catalytic activity than either mutation alone. Furthermore, we hypothesized that the overall three-dimensional structure of the protein will not be grossly altered by combining these two subtle mutations. Therefore, we have constructed this novel double mutant, which we have named R338A + FIXVIIEGF1, and have evaluated it for hemophilia B gene therapy.
In terms of gene therapy, expression of a catalytically enhanced FIX molecule from a vector would be highly beneficial because it would permit administration of lower vector doses to achieve the same therapeutic effect compared with the same vector expressing the wild-type molecule, thus increasing the therapeutic index. Likewise, for protein replacement therapy, lower amounts of the catalytically enhanced FIX molecules would be required for therapeutic benefit compared with the wild-type molecule, thus potentially reducing the cost significantly to perhaps permit the more desirable and effective prophylactic treatment regimen. In this study, we have evaluated the gene therapy potential of these three FIX variants in the context of the helper-dependent adenoviral vector (HDAd). HDAds are devoid of all viral sequences and thus are attractive for hemophilia B gene therapy because they can mediate long-term transgene expression in hemophilia B mice (Ehrhardt and Kay, 2002) and dogs (Brunetti-Pierri et al., 2005a) without chronic toxicity.
Materials and Methods
Generation of bioengineered hFIX
Oligonucleotides 5′-GAC CGA GCC ACA TGT CTT GCA TCT ACA AAG TTC ACC ATC-3′ and 5′-GAT GGT GAA CTT TGT AGA TGC AAG ACA TGT GGC TCG GTC-3′ were used to generate the arginine-to-alanine modification in R338A-FIX and R338A + FIXVIIEGF1, using a QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Oligonucleotides 5′-GTG ACC AGT GTG CCT CAA GTC CAT GCC AGA ATG GGG GCT CCT GCA AGG ACC AGC TCC AGT CCT ATA TCT GCT TCT GCC TCC CTG CCT TCG AGG GCC GGA ACT GTG-3′ and 5′-AGC TCA CAG TTC CGG CCC TCG AAG GCA GGG AGG CAG AAG CAG ATA TAG GAC TGG AGC TGG TCC TTG CAG GAG CCC CCA TTC TGG CAT GGA CTT GAG GCA CAC TG-3′ containing the human FVII EGF-1 domain were hybridized, and used to replace the BstEII–SacI fragment containing the endogenous EGF-1 domain of human FIX to generate the FIXVIIEGF1 modification. All cDNAs were sequenced to verify the desired modification.
Helper-dependent adenoviral vectors
All vectors were constructed in the pΔ21.7E4 backbone (Brunetti-Pierri et al., 2005b) and they all contained an expression cassette composed of the following elements (from 5′ to 3′): a liver-restricted rat phosphoenolpyruvate carboxykinase (PEPCK) promoter (Beale et al., 1992), the apolipoprotein AI (ApoAI) intron, the human FIX cDNA, the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), the apolipoprotein E (ApoE) locus control region (LCR), and the human growth hormone polyadenylation signal sequence [p(A)]. HDAds were produced in 116 cells (Palmer and Ng, 2003) with the helper virus AdNG163 (Palmer and Ng, 2004) as described in detail elsewhere (Palmer and Ng, 2003). Helper virus contamination levels were determined as described elsewhere (Palmer and Ng, 2003) and were found to be <0.05% for all vector preparations. DNA analyses of HDAd genomic structure were confirmed for both vectors as described elsewhere (Palmer and Ng, 2003).
Animal experiments
Animal procedures were approved by the Institutional Animal Care and Use Committee. Animals used for the experiments included the following: hemophilia B (FIX knockout) mice (Wang et al., 1997) and C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). For intravenous vector administration, retro-orbital injection was performed in hemophilia B mice (8–12 weeks old) at a dose of 1 × 1012 vector particles (VP)/kg. For the studies assessing thrombotic risk, C57BL/6 mice were injected at a dose of 5 × 1012 VP/kg. Statistical analyses were performed with the t test.
FIX antigen and FIX activity
Plasma human FIX antigen levels were measured by enzyme-linked immunosorbent assay (ELISA) (Enzyme Research Laboratories, South Bend, IN) according to the manufacturer's protocol. FIX activity was measured by the one-stage clotting assay, using human FIX-deficient plasma. The percentage of activity in plasma was determined from a calibration curve prepared with pooled plasma from C57BL/6 mice (n = 5). The aPTT reagent used was STA PTT Automate 5 (Diagnostica Stago, Parsippany, NJ) and was measured with an STart 8 hemostasis analyzer (Diagnostica Stago). FIX-deficient plasma was purchased from Precision BioLogic (Dartmouth, NS, Canada).
Markers of thrombus formation
Quantitative assessment of D-dimer levels in plasma was performed by an immunoturbidimetric method (Diagnostica Stago). In this method, a beam of monochromatic light is allowed to traverse a suspension of microlatex particles to which specific antibodies have been attached by covalent bonding. In the presence of D-dimers, the antibody-coated latex particles agglutinate to form aggregates causing an increase in light absorbance. This increase in light absorption is a function of the antigen level present in the test sample. Using chromogenic methodology (Diagnostica Stago), quantitative antithrombin levels are measured by a two-step procedure: first, the plasma is incubated with a known excess of thrombin in the presence of heparin; second, the residual thrombin is quantitated by its amidolytic action on the synthetic chromogenic substrate CBS 61.50. The antithrombin level of the tested sample is inversely proportional to the residual thrombin in the second reaction step.
Results
Factor IX antigen levels and duration of expression
Hemophilia B mice (n = 5 or 6) were injected with HDAd expressing the bioengineered FIX molecules or the wild-type FIX at a dose of 1 × 1012 VP/kg. Two, four, thirty-six, and forty-four weeks postinjection, the levels of plasma FIX antigen were determined for each mouse (Fig. 3). Injection of all four vectors resulted in stable, high levels of FIX (Fig. 3). These results indicate that the bioengineered FIX variants do not compromise the duration of expression, suggesting that they are no more immunogenic than the wild-type FIX, at least in C57BL/6 hemophilia B mice.
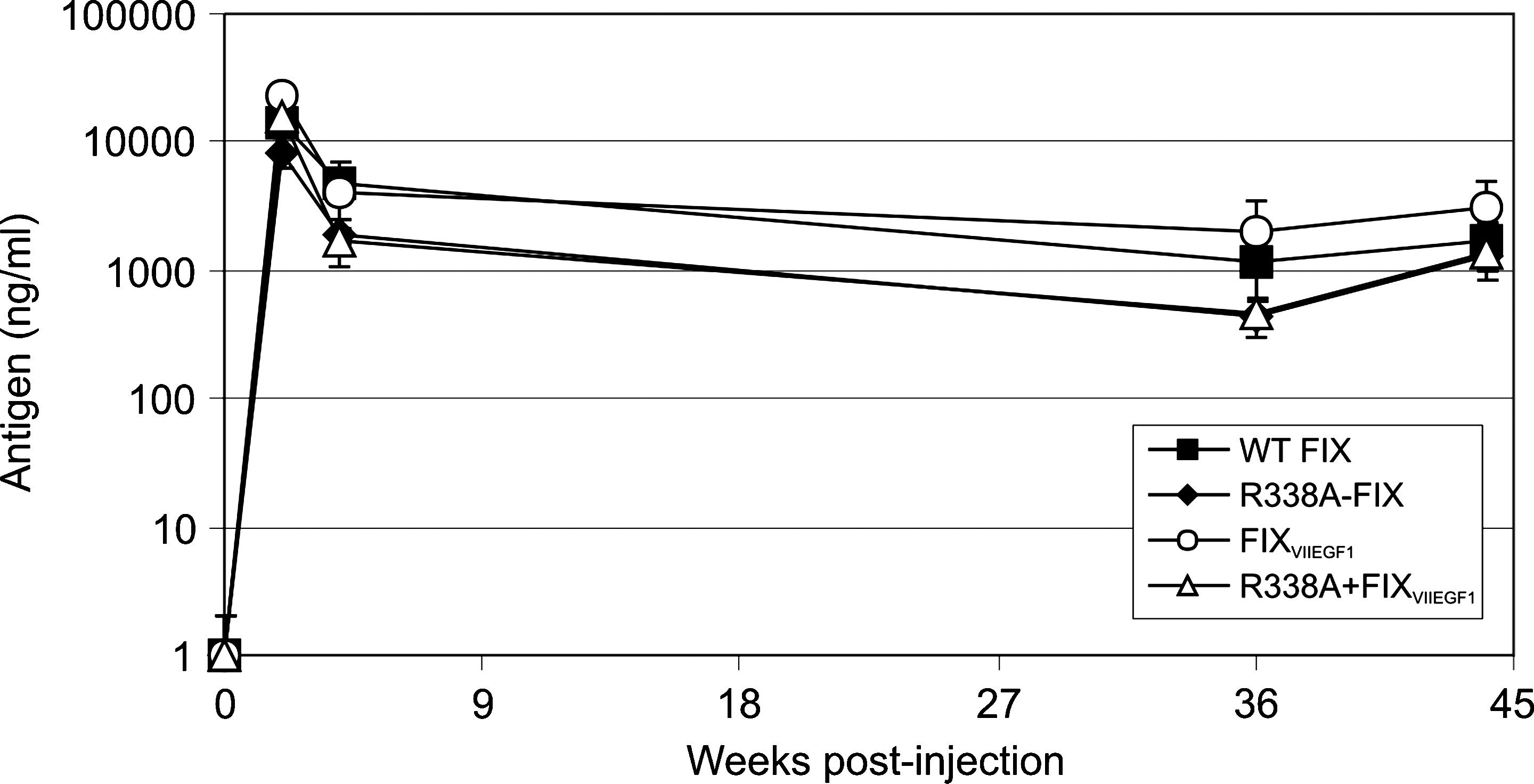
Duration of FIX antigen expression in hemophilia B mice after intravenous administration of HDAd (1 × 1012 VP/kg) encoding wild-type FIX, R338A-FIX, FIXVIIEGF1, or R338A + FIXVIIEGF1.
Specific activity
We hypothesized that for a given dose, vectors expressing the bioengineered FIX molecules (R338A-FIX, FIXVIIEGF1, and R338A + FIXVIIEGF1) will result in higher specific activities (IU/mg) than the vector expressing the wild-type molecule. To test this, the percentage of normal FIX activity was determined for each of the aforementioned injected mice and the specific activity (IU/mg) was calculated to permit direct comparison between the four different treatment groups. By comparing specific activity, variables such as slight differences in vector infectivity (which may account for the differences in FIX antigen levels despite identical vector doses) or the exact amount of plasma antigen can be eliminated. The results indicated that compared with the vector expressing wild-type human FIX, the vector expressing R338A-FIX was more efficacious, yielding 2.2-fold higher activity at 2 weeks (p = 0.00039), 3.3-fold higher at 4 weeks (p = 9.7 × 10–6), 3.6-fold higher at 36 weeks (p = 1.1 × 10–5), and 2.3-fold higher at 44 weeks (p = 0.0012) than FIX-specific activities from the vector expressing the wild-type FIX molecule (Fig. 4), for an overall average increase of 2.9 ± 0.7-fold over wild-type. In the case of the vector expressing FIXVIIEGF1 the specific activity was 2.0-fold higher at 2 weeks (p = 0.00032), 4.8-fold higher at 4 weeks (p = 5.8 × 10–7), 3.7-fold higher at 36 weeks (p = 0.0025), and 3.3-fold higher at 44 weeks (p = 0.00015) than that of wild-type FIX (Fig. 4) for an overall average increase of 3.4 ± 1.1-fold over wild type. In the case of the vector expressing the novel double modification, R338A + FIXVIIEGF1, the specific activity was 8.9-fold higher at 2 weeks (p = 1.4 × 10–9), 19.9-fold higher at 4 weeks (p = 5.8 × 10–6), 16.7-fold higher at 36 weeks (p = 4.3 × 10–6), and 8.1-fold higher at 44 weeks (p = 6.8 × 10–6) than that of wild-type FIX (Fig. 4) for an overall average increase of 12.6 ± 4.8-fold over wild-type. These results confirm that the single modifications (R338A and FIXVIIEGF1) have significantly higher catalytic activity than the wild-type and suggest that they may improve the safety and efficacy of HDAd by permitting the use of lower vector doses to achieve the same therapeutic outcome. Furthermore, these results also indicate that the novel double modification (R338A + FIXVIIEGF1) has an even higher catalytic activity than each single mutant alone and may thus increase the therapeutic index of the vector even further.
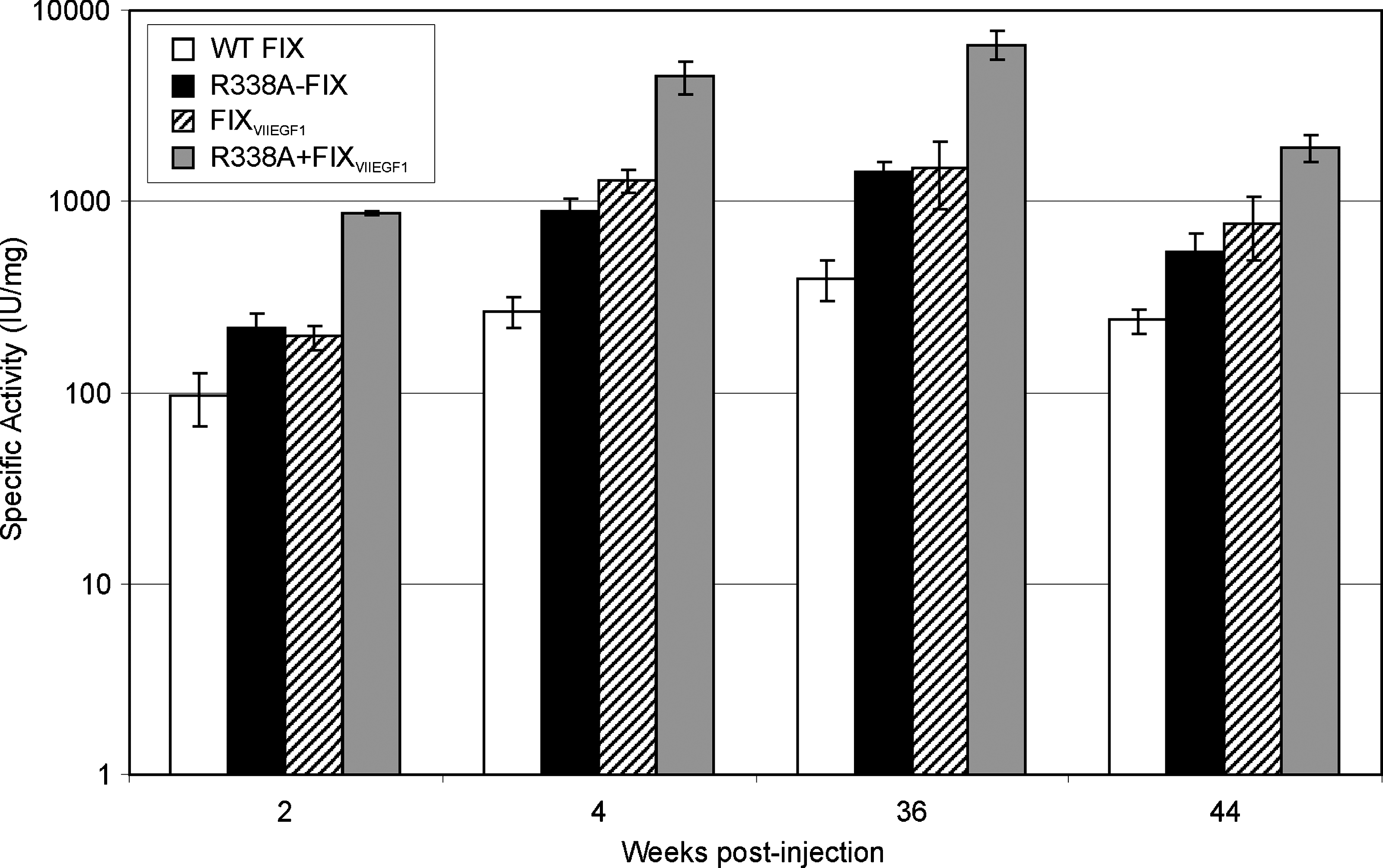
Specific activity (IU/mg) in hemophilia B mice 2, 4, 36, and 44 weeks after intravenous administration of HDAd (1 × 1012 VP/kg) encoding wild-type FIX, R338A-FIX, FIXVIIEGF1, or R338A + FIXVIIEGF1. Shown are averages ± SD (n ≥ 5).
To confirm this, hemophilia B mice (n = 3 or 4 per group) were injected with a vector encoding either the wild-type FIX or R338A + FIXVIIEGF1 at a 10-fold lower dose (1 × 1011 VP/kg) as compared with the previous experiment. Two weeks postinjection, the plasma FIX antigen level and the percentage of normal FIX activity were determined. Mice injected with the vector encoding R338A + FIXVIIEGF1 had 218.7 ± 30.7% of normal FIX activity (2.2 IU/ml) whereas mice injected with the vector encoding wild-type FIX had only 41.8 ± 5.9% normal FIX activity (0.42 IU/ml) (Fig. 5A). This 5.2-fold greater FIX activity (p = 8.4 × 10–5) was achieved despite the fact that the amount of FIX antigen was 1.6-fold higher (p = 0.033) in the mice injected with the vector expressing wild-type FIX (1192 ± 242 ng/ml) compared with the mice injected with vector encoding R338A + FIXVIIEGF1 (742.7 ± 112.7 ng/ml) (Fig. 5B). Thus, these results confirm that greater therapeutic benefit can be obtained with a lower dose of the vector expressing R338A + FIXVIIEGF1 compared with wild-type FIX.
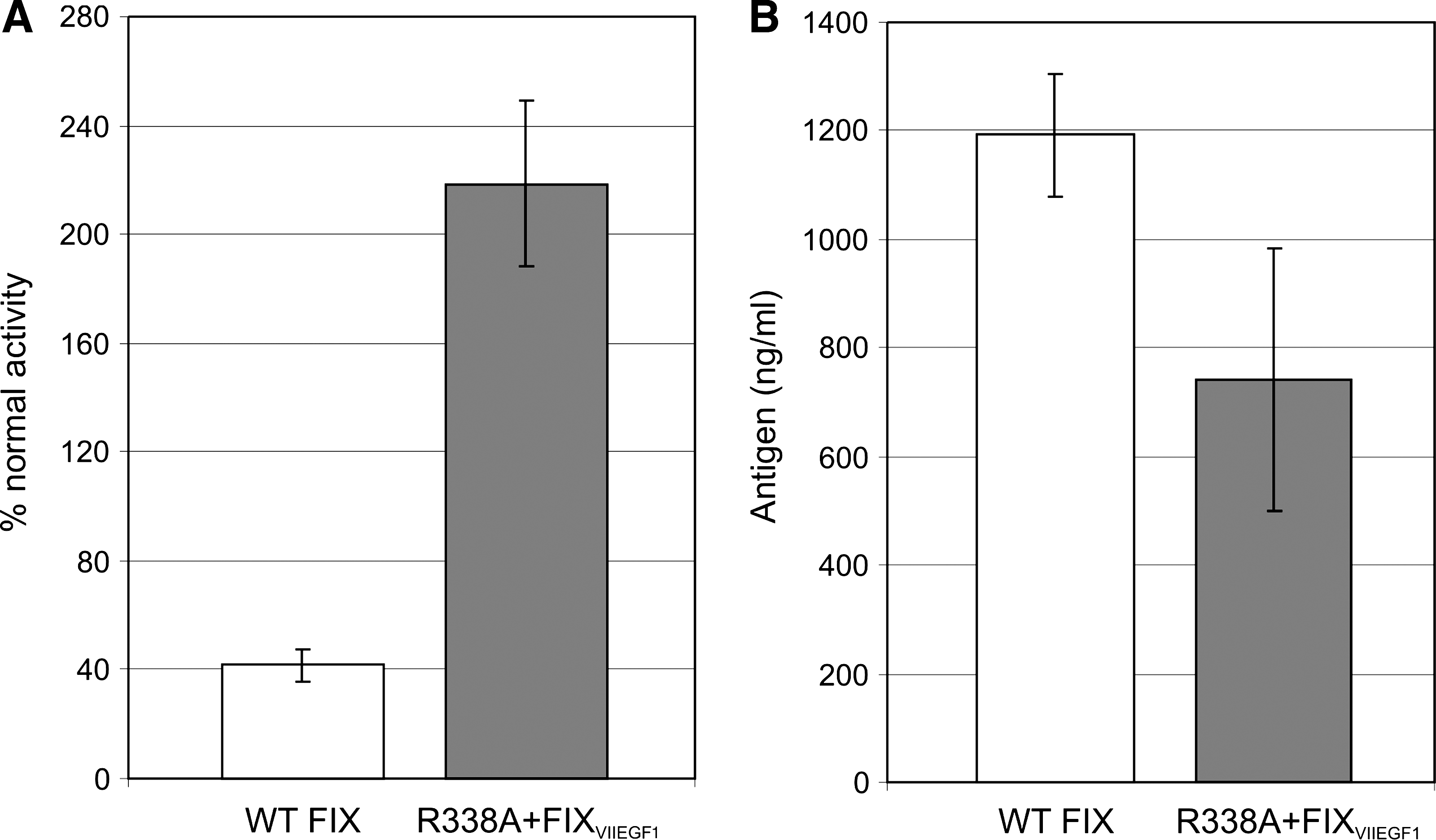
(
Thrombogenicity
One concern regarding the use of catalytically enhanced FIX molecules is their potential to increase the risk for thrombotic complications. To assess for hypercoagulable state, we measured antithrombin III, fibrinogen, and D-dimer levels in wild-type mice (n = 3) injected with HDAd (5 × 1012 VP/kg) expressing either wild-type FIX or one of the three bioengineered FIX variants. The higher dose was chosen to magnify any thrombogenicity if present. Despite stable expression of supraphysiological levels of FIX for more than 1 year (data not shown), there were no significant differences (p ≥ 0.12) in any of the three aforementioned parameters (Table 1), suggesting that the three bioengineered molecules were no more thrombogenic than the wild-type molecule.
Abbreviation: FIX, factor IX; HDAd, helper-dependent adenovirus.
HDAd (5 × 1012 VP/kg) expressing the indicated FIX variant was injected into wild-type mice (n = 3). Means ± SD are shown.
Measured 3 weeks after injection of vector.
Measured 2 weeks after injection of vector.
Discussion
In this study, we have sought to increase the safety and efficacy of hemophilia B gene therapy by using gene transfer vectors that express catalytically enhanced FIX molecules, with the rationale that such variants would increase the vector therapeutic index by permitting therapeutic outcome with lower doses. This is important because vector-mediated toxicity is dose dependent (Brunetti-Pierri et al., 2004). We have evaluated vectors expressing two previously described bioengineered FIX molecules, R338A-FIX and FIXVIIEGF1, and indeed have found 2.9- and 3.4-fold increases in specific activity, respectively, compared with a vector that expressed wild-type FIX, after vector administration to hemophilia B mice. These levels of enhancement are consistent with those observed for the purified, recombinant bioengineered molecules (Chang et al., 1997, 1998). R338A-FIX has been used in a gene therapy setting with AAV (Schuettrumpf et al., 2005). In that study, a 6-fold enhancement in FIX-specific activity over the wild-type FIX was reported after intravenous injection. We do not understand why there is a difference in the fold increase in FIX-specific activity among the R338A-FIX molecules in our study and in the study by Schuettrumpf and colleagues (2005); however, it should be noted that the study that originally reported the R338A variant (Chang et al., 1998) showed approximately three times greater clotting activity than the wild-type FIX, which is closer to our results than the results reported by Schuettrumpf and colleagues (2005). Regardless, we believe the difference is modest (less than an order of magnitude). In an effort to further increase the therapeutic index, we produced and evaluated a novel FIX variant that combined R338A-FIX and FIXVIIEGF1 into a single molecule and have found a 12.6-fold increase in specific FIX activity. The increase in catalytic activity was confirmed with a lower vector dose, thus confirming that the double modification in the FIX molecule allows the use of a lower vector dose to achieve improved therapeutic outcome (Fig. 5B). The use of bioengineered FIX molecules with enhanced specific activity to treat hemophilia B patients would offer many advantages. In the gene therapy context, use of such molecules would greatly improve safety and efficacy by permitting the administration of lower vector doses to achieve therapeutic benefit. For example, the innate inflammatory immune response to Ad-based vectors is dose dependent, exhibiting a steep threshold effect (Morral et al., 2002; Brunetti-Pierri et al., 2004). Therefore, any reduction in vector dose would greatly increase the vector therapeutic index. In the case of protein replacement therapy, reduced amounts of the bioengineered FIX molecules would be required to achieve hemostasis compared with the wild-type molecule, thus potentially reducing the patient's cost significantly and perhaps permitting a more desirable and effective prophylactic treatment regimen.
One concern regarding the use of catalytically enhanced FIX is the risk of increased thrombosis. However, the evaluation of antithrombin III, fibrinogen, and D-dimer revealed that the three variants were no more thrombogenic than the wild-type molecule, even when present at supraphysiological levels. Thus, we conclude that the three bioengineered FIX molecules are no more thrombogenic than wild-type FIX. This is perhaps not unexpected because the modifications of the three variants do not alter normal FIX regulation (Chang et al., 1997, 1998). Consistent with our results, purified FIXVIIEGF1 was not observed to result in any thrombotic complications when administered to hemophilia B dogs (Chang et al., 1997).
Hemophilia B patients develop inhibitors (neutralizing anti-FIX antibodies) at a frequency of 1–3%, which is detrimental because exogenously administered FIX protein will no longer be effective in response to bleeds. Therefore, another concern with bioengineered FIX variants involves the potential for increased risk of inhibitor formation. However, expression of all three variants, like the wild-type, persisted for the duration of the observation period of at least 44 weeks at a dose of 1 × 1012 VP/kg and more than 1 year at a dose of 5 × 1012 VP/kg, indicating that the variants are no more immunogenic than the wild-type molecule, at least in C57BL/6 hemophilia B mice.
In summary, we demonstrated that intravenous delivery of HDAd encoding R338A, FIXVIIEGF1, and R338A + FIXVIIEGF1 resulted in circulating levels of FIX with higher biological activity compared with wild-type FIX. These bioengineered FIX molecules, and in particular our novel R338A + FIXVIIEGF1 variant, may improve the safety and efficacy of HDAd by permitting the use of lower vector doses to achieve the same therapeutic outcome. In addition, these bioengineered FIX molecules may be valuable for other vector systems as well as for recombinant FIX protein replacement therapy. In the hemophilia B liver-directed clinical trial with AAV vector, the fact that two subjects developed transient transaminitis suggests a direct relationship between the AAV dose and the levels of hepatotoxicity as a consequence of an adaptive cellular immune response against the transduced hepatocytes (Manno et al., 2006; Mingozzi et al., 2007). It has been suggested that reducing the AAV dose may help to evade this immune response (Mingozzi and High, 2007). Therefore, the bioengineered FIX variants with increased catalytic activity described herein may be useful for achieving therapeutic FIX activity with lower vector doses.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health (R01 DK069369 to P.N.; K99HL088692 to N.B.-P.) and the Texas Affiliate of the American Heart Association (0765032Y to N.B.-P.). The financial support of Telethon-Italy (Fellowship GFP04008) to N.B.-P. is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.