
Abstract
A major obstacle for the efficacy of cancer gene therapy is the need to transduce a high proportion of tumor cells with genes that directly or indirectly cause their death. During the formation of certain organs, cells compete among themselves to colonize the whole tissue. We reasoned that cell competition could be used to increase the proportion of cells that become transfected in a tumor. For this, a transgene that provides a selective advantage to the transfected cells should be used. If the same gene conferred a suicide mechanism the tumor could be eradicated after a period of selection. Bystander effect of transfected cells over neighboring nonmodified cells may eliminate tumors even with incomplete replacement of tumor cells. To test this strategy a competitive advantage was provided to colon cancer cells, using a gene encoding a fusion protein of dihydrofolate reductase (DHFR) and thymidine kinase (TK). DHFR confers resistance to methotrexate (MTX) and TK confers sensitivity to ganciclovir (GCV). Modified cells were also transduced with green fluorescent protein and parental cells with red fluorescent protein. In vitro and in vivo experiments were performed, using various proportions of modified cells and applying positive selection with MTX followed by negative selection with GCV. In vitro, cell competition was evident. Under MTX treatment, tumor cells transfected with the DHFR–TK fusion gene efficiently replaced the parental cells (from 0.1 to 90% in 35 days). After this positive selection period, negative selection with GCV eliminated the transfected cells. In vivo, positive selection was also achieved and resulted in a statistically significant therapeutic effect.
Introduction
I
It is well known that during the formation of certain organs, cells compete among themselves to colonize the whole tissue (Moreno et al., 2002; Moreno and Basler, 2004; Diaz and Moreno, 2005). During chemotherapy treatment there is selective pressure inside the tumor that results in expansion of resistant cells (Fig. 1A) (de Anta et al., 2006). The search for genes that can be selected in vivo has been a goal in chemoprotective gene therapy. Chemoprotection seeks to protect bone marrow and other organs from adverse effects of high-dose chemotherapy (Banerjee et al., 1994; Licht et al., 1997). The genes most commonly used have been dihydrofolate reductase (DHFR) (Bertino et al., 1996) and multidrug resistance (MDR1) genes (Bosch and Croop, 1996). DHFR is the cellular enzyme that regenerates the tetrafolate used during the conversion of dUMP to TMP in the synthesis of nucleotides. DHFR can be inhibited by MTX, which results in the blockade of the synthesis of purines, thymidylate, and certain amino acids. MTX is used to treat acute lymphoblastic leukemia and lymphoma, but it is highly myelosuppressive. DHFR mutants with highly reduced binding affinity for MTX, such as the Phe22–Ser31 double mutant DHFR (dmDHFR) (Ercikan-Abali et al., 1996; Sauerbrey et al., 1999), can protect bone marrow from MTX-induced myelosuppression and have been used to positively select transfected hematopoietic cells in vivo (Meisel et al., 2003). As an initial approach toward positive selection of transfected cancer cells in vivo we decided to use this dmDHFR encoded in an episomal plasmid. This mechanism maintains protein expression during transfected cell propagation. In the context of negative selection with HSV-TK and GCV, the use of DHFR has an additional advantage. Coexpression of TK and DHFR potentiates the resistance to MTX because they contribute to bypass the block of thymidylate synthesis (Mineishi et al., 1997). Furthermore, the group lead by J. Bertino has demonstrated that a fusion protein of DHFR and HSV-TK preserves the activity of both proteins and is overexpressed in the presence of MTX (Mayer-Kuckuk et al., 2002). This upregulation of HSV-TK expression by means of fusion to DHFR and administration of MTX in vivo has been proposed as a way to increase the expression level of TK in tumor cells (Mayer-Kuckuk et al., 2003). We have therefore used a similar construct fusing dmDHFR and HSV-TK in order to enrich the transfected cell population of a tumor by means of MTX administration. Transfected cell enrichment should be associated with an increased GCV therapeutic effect, and in a model with bystander effect, the enrichment of modified cells could eliminate tumors after suicide-inducing prodrug treatment (a scheme is presented in Fig. 1B). In the absence of a bystander effect natural drug-resistant cells would survive after GCV treatment, and therefore the bystander effect is important to eliminate all tumor cells completely. A similar enrichment–suicide approach has been proposed, using the FCU1 gene and N-(phosphonacetyl)-
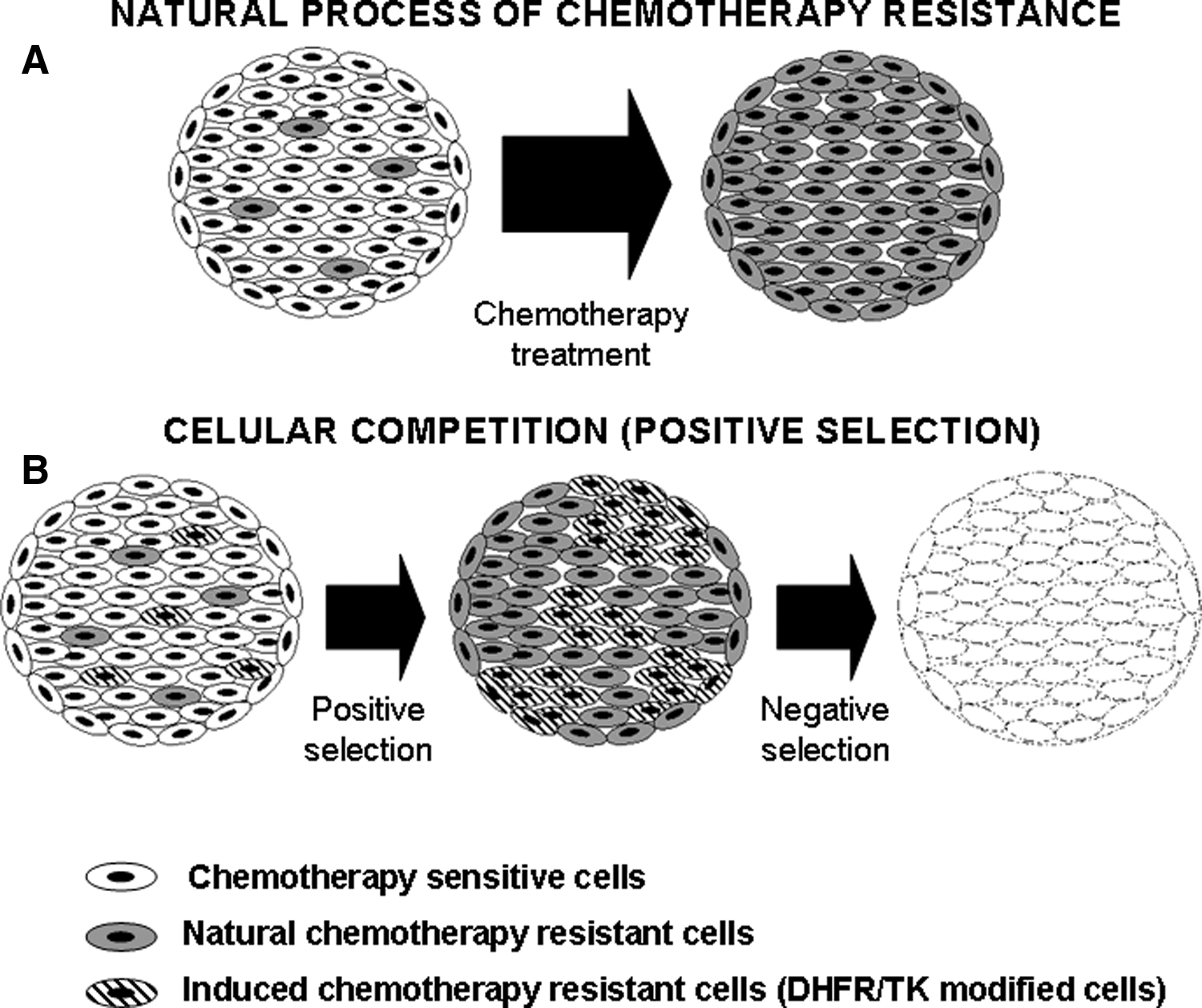
Natural process of chemotherapy resistance compared with the proposed strategy of cell competition. (
Materials and Methods
Cell culture and reagents
The human colon cancer cell line HCT116 was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin (P/S). Cells were maintained at 37.0°C and 5% CO2 in a humidified atmosphere and were mycoplasma negative. Methotrexate (MTX) was provided by Wyeth Pharma (Madrid, Spain). A 54 mM stock solution was prepared in phosphate-buffered saline (PBS) and dilutions were made in PBS. Ganciclovir (GCV) was provided by Roche Diagnostics (Mannheim, Germany). A stock solution at 100 mg/ml was prepared in PBS and dilutions were made in PBS.
Plasmid construction
The green fluorescent protein (GFP) gene from pEPI-1 (Jenke et al., 2002) was replaced with a TK–EGFP fusion gene from plasmid pTK-EGFP (Cascante et al., 2005). The resulting plasmid, pEPI-TKGFP, contains the TK–GFP fusion gene driven by the cytomegalovirus (CMV) promoter. It also contains a chromosomal scaffold/matrix-attached region and the simian virus 40 (SV40) origin of replication to achieve episomal propagation in eukaryotic cells (Jenke et al., 2002). We synthesized a double mutant Phe22–Ser31 DHFR gene (Sauerbrey et al., 1999) using 24 overlapping oligonucleotides spanning the 564 base pairs of the DHFR cDNA plus appropriate restriction sites at both ends and cloned it into pBluescript to obtain pBS-dmDHFR. The GFP gene from pEPI-TKGFP was replaced with the dmDHFR gene to obtain pEPI-DHFR/TK.
Generation of stable HCT116DHFR/TK-GFP and HCT116-RFP clones
HCT116 cells were seeded in a 6-well plate and transfected 24 hr later with 3 μg of pEPI-DHFR/TK plasmid with FuGENE 6 transfection reagent (Roche Diagnostics) according to manufacturer protocols. HCT116DHFR/TK stable clones were selected with 0.625 μM MTX for 24 days. We used red fluorescent protein (RFP) and green fluorescent protein (GFP) as marker genes of the parental and transfected cells, respectively. The packaging cell lines PA317DsRED and PA317DsGFP (kindly provided by F. Larcher, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas, CIEMAT, Madrid, Spain) were used to produce Moloney leukemia vector RED2 and Moloney leukemia vector GFP, respectively. Cells were grown in DMEM–10% FBS and medium was replaced when cells reached 70–80% of confluence. PA317DsRED and PA317DsGFP conditioned medium was used to infect HCT116 or HCT116DHFR/TK, respectively, in the presence of Polybrene (8 μg/ml; Sigma-Aldrich, St. Louis) and DMEM–5% FBS. Twelve hours after infection, medium was replaced with fresh medium and cells were seeded in a 96-well plate at a confluence of two or three cells per well. After incubation for 21 days and under fluorescence microscopy we obtained HCT116-RFP and HCT116DHFR/TK-GFP clones.
In vitro MTX and GCV sensitivity of HCT116DHFR/TK-GFP and HCT116 RFP cells
HCT116-RPF and HCT116DHFR/TK-GFP cells were plated in 96-well plates (2.5 × 104 cells per well). Two days later, cells were incubated with various concentrations of MTX or GCV. Cell viability was measured by bicinchoninic acid (BCA) protein assay (Pierce Biotechnology/Thermo Fisher Scientific, Rockford, IL) 12 days after treatment. The percentage of cell viability was defined by the equation T/NT × 100, where T is the viability of treated cells and NT is the viability of nontreated cells.
In vitro positive and negative selection
A mixture of 0.1% HCT116DHFR/TK-GFP cells and 99.9% parental HCT116-RFP cells was seeded in quadruplicate in 3.5-cm plates (1.3 × 106 cells per plate). Two days later, cells were incubated with 0.625 μM MTX. The selection medium was replaced three times every week. Images (×100) were taken with a confocal microscope (spectral confocal TCS-SL; Leica Microsystems, Wetzlar, Germany) on various days after treatment with MTX. To quantify the amount of GFP and RFP cells, cell monolayers were harvested on various days after MTX administration and cells were fixed with 4% paraformaldehyde (PFA) in PBS overnight. After washing with PBS, cells were resuspended at 106 cells/ml PBS and at least 50,000 cells were analyzed by flow cytometry (FACSCalibur system; BD Biosciences, San Jose, CA) using 488- and 633-nm lasers and CellQuest Pro software.
For negative selection, after in vitro MTX treatment, cells were incubated with GCV (10 μg/ml) for 20 days (MTX was also maintained). Confocal microscopy images (×100) were taken on various days during the treatment. To quantify the degree of negative selection, cells were harvested and analyzed by flow cytometry according to the previously described protocol.
In vivo positive selection
To allow for positive selection, subcutaneous xenografts were established in the flanks of 4- to 8-week-old male BALB/c nu/nu mice by inoculation of a mixture of 3.5 × 106 cells consisting of 1% HCT116DHFR/TK-GFP cells and 99% parental HCT116-RFP cells. Animals were kept and manipulated in accordance with recommendations of the Federation of European Laboratory Animal Science Associations for the proper use of laboratory animals. Fifteen days later, animals received MTX (9 mg/kg) or PBS intraperitoneally three times every week. On the indicated day of treatment (day 0, 10, 17, 24, or 31) animals were killed and tumors were excised and fixed with 4% paraformaldehyde (PFA) in PBS overnight. After washing with PBS, tumors were incubated with sucrose 5% in PBS (2 hr) and sucrose 30% in PBS (overnight). Tumors were then cryopreserved with O.C.T. medium and cryosectioned at 5 μm with a cryotome (Shandon cryotome cryostat; Global Medical Instrumentation, Ramsey, MN). Sections were mounted and GFP and RFP expression was viewed with a confocal microscope. To compensate for heterogeneity within the tumors, three images (×400) were taken from the same section. Quantification was performed by a blinded observer and three fields of each section were counted.
In vivo bystander effect of HCT116DHFR/TK-GFP cells with GCV
Subcutaneous xenografts were established in the flanks of 4- to 8-week-old male BALB/c nu/nu mice by inoculation of 3.5 × 106 cells consisting of 100% HCT116-RFP cells, 100% HCT116DHFR/TK-GFP cells, or a mixture of 50% HCT116DHFR/TK-GFP cells and 50% of HCT116-RFP cells. Once tumors reached 100–170 mm3, animals started GCV treatment (75 mg/kg, administered intraperitoneally daily for 14 days) (n = 6–8). Tumors were measured two times weekly and tumor volume was calculated according to the equation
In vivo positive and negative selection
The positive and negative selection strategy was performed using two different proportions of HCT116DHFR/TK-GFP cells. In one experimental setting subcutaneous xenografts were established in the flanks of 4- to 8-week-old male BALB/c nu/nu mice by inoculation of a mixture of 3.5 × 106 cells consisting of 1% HCT116DHFR/TK-GFP cells and 99% parental HCT116-RFP cells. One day later, animals received MTX (9 mg/kg) or PBS intraperitoneally three times per week. Once tumors reached 300 mm3, animals finished MTX treatment (positive selection) and were treated intraperitoneally with GCV (50 mg/kg) daily for 14 days (negative selection) (n = 9). In a different experimental setting subcutaneous xenografts were established in the flanks of 4- to 8-week-old male BALB/c nu/nu mice by inoculation of a mixture of 3.5 × 106 cells consisting of 6% HCT116DHFR/TK-GFP cells with 94% parental HCT116-RFP cells. One day later, animals were randomly distributed into four groups (MTX+PBS, MTX+GCV, PBS+PBS, and PBS+GCV). The positive selection phase (treatment with MTX or PBS) was performed from 1 day after tumor implantation until tumors reached 100–170 mm3. When we finished positive selection we began intraperitoneal GCV treatment (75 mg/kg) daily for 14 days (n = 9). Tumors were measured two times weekly.
Results
In vitro MTX and GCV sensibility of HCT116DHFR/TK-GFP and HCT116-RFP cells
We constructed a DHFR/TK fusion gene to allow for positive–negative selection of transfected cells. As a plasmid backbone we used pEPI-1, a plasmid that contains a eukaryotic origin of replication and chromosomal elements for replication and segregation. The resulting plasmid, pEPI-DHFR/TK, was transfected into HCT116 colon carcinoma cells, which were then selected for a population of MTX-resistant cells. After MTX selection, resistant cells were infected with green fluorescent protein recombinant retrovirus, and an HCT116DHFR/TK-GFP clone was selected. In parallel, parental HCT116 cells were infected with a red fluorescent protein recombinant retrovirus and a parental HCT116-RFP clone was selected. We determined the in vitro sensitivity to MTX and GCV of the selected HCT116DHFR/TK-GFP and HCT116-RFP clones. The MTX median inhibitory concentration (IC50) for HCT116DHFR/TK-GFP cells was about 5 μM whereas for the parental HCT116-RFP cells the MTX IC50 was less than 0.625 μM (Fig. 2A). These results indicate that under MTX, HCT116DHFR/TK-GFP cells have a proliferative advantage with respect to HCT116-RFP cells (positive selection). On the other hand, HCT116DHFR/TK-GFP cells were sensitive to GCV doses as low as 0.625 μg/ml (Fig. 2B). These results indicate that HCT116DHFR/TK-GFP cells could be used for positive selection with MTX and negative selection with GCV.

HCT116DHFR/TK cells are resistant to MTX and sensitive to GCV in vitro. HCT116-RFP cells (open columns) and HCT116DHFR/TK-GFP cells (solid columns) were plated in 96-well plates. Two days later, cells were incubated with various concentrations of (
In vitro positive selection with MTX
Next we determined whether a small proportion of HCT116DHFR/TK-GFP cells could compete out parental HCT116-RFP cells under MTX treatment. After 15 days of MTX treatment, 0.1% of HCT116DHFR/TK-GFP cells overgrew HCT116-RFP cells. Without MTX treatment the transfected cells did not show any advantage over parental cells (Fig. 3A). To verify the results of positive selection quantitatively, we analyzed the samples by flow cytometry and the results demonstrated that under MTX treatment, the proportion of HCT116DHFR/TK-GFP cells increased from 0.1 to 90% in 35 days (Fig. 3B).
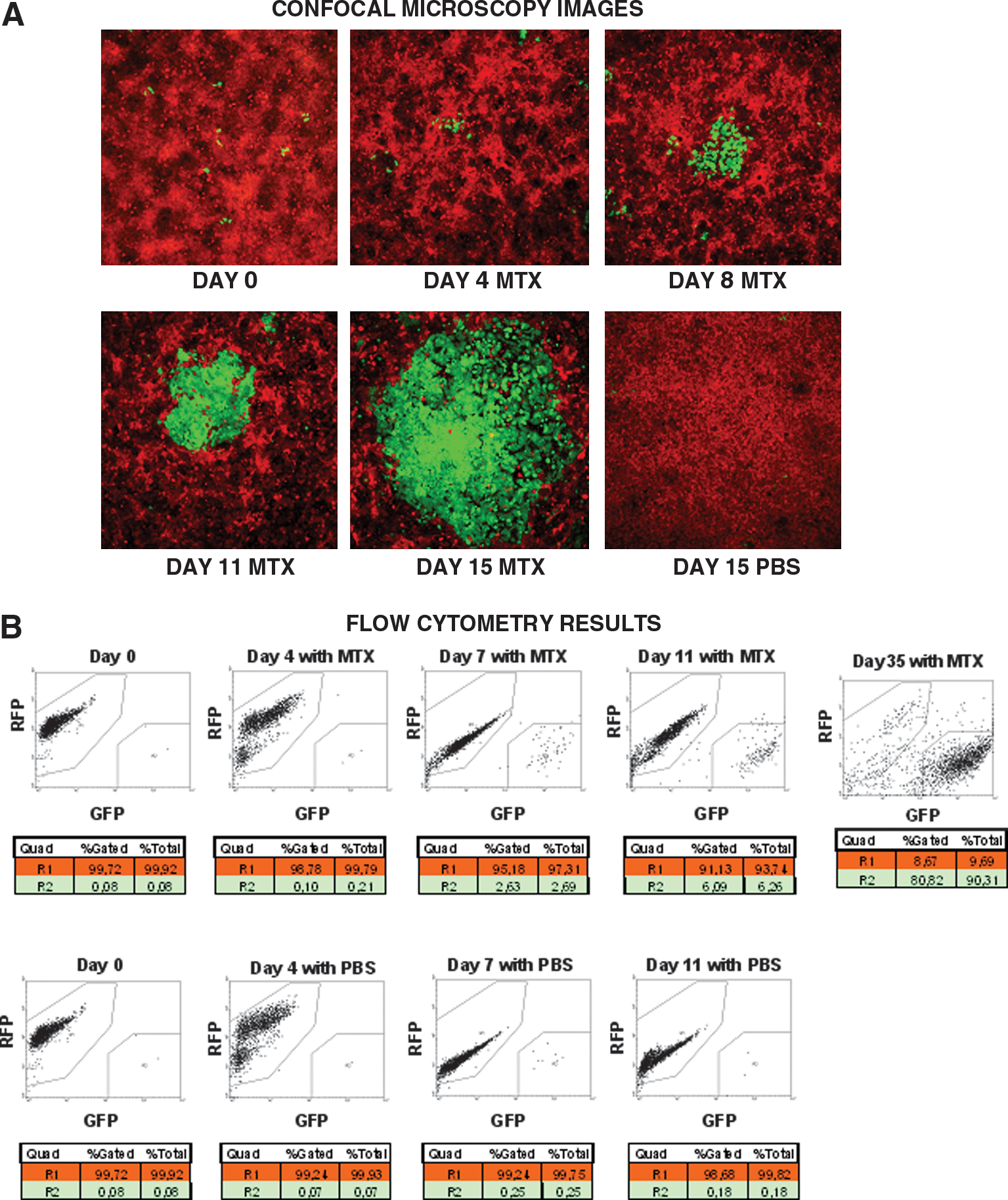
DHFR/TK-transfected cells (green) outcompete parental cells (red) in vitro. (
In vitro negative selection with GCV
We then examined how positive selection with MTX followed by negative selection with GCV could help to eradicate all the cells in a monolayer culture in vitro. After 15 days of MTX positive selection HCT116DHFR/TK-GFP cells were eliminated with GCV but nontransfected HCT116-RFP cells survived, indicating a poor bystander effect. All the areas corresponding to HCT116DHFR/TK-GFP cells were cleared from the cultures, but the adjacent areas of parental cells remained unaltered (Fig. 4A). To verify the results of negative selection quantitatively, we analyzed the samples by flow cytometry and the results confirm that on GCV addition only transfected cells were eliminated (Fig. 4B).
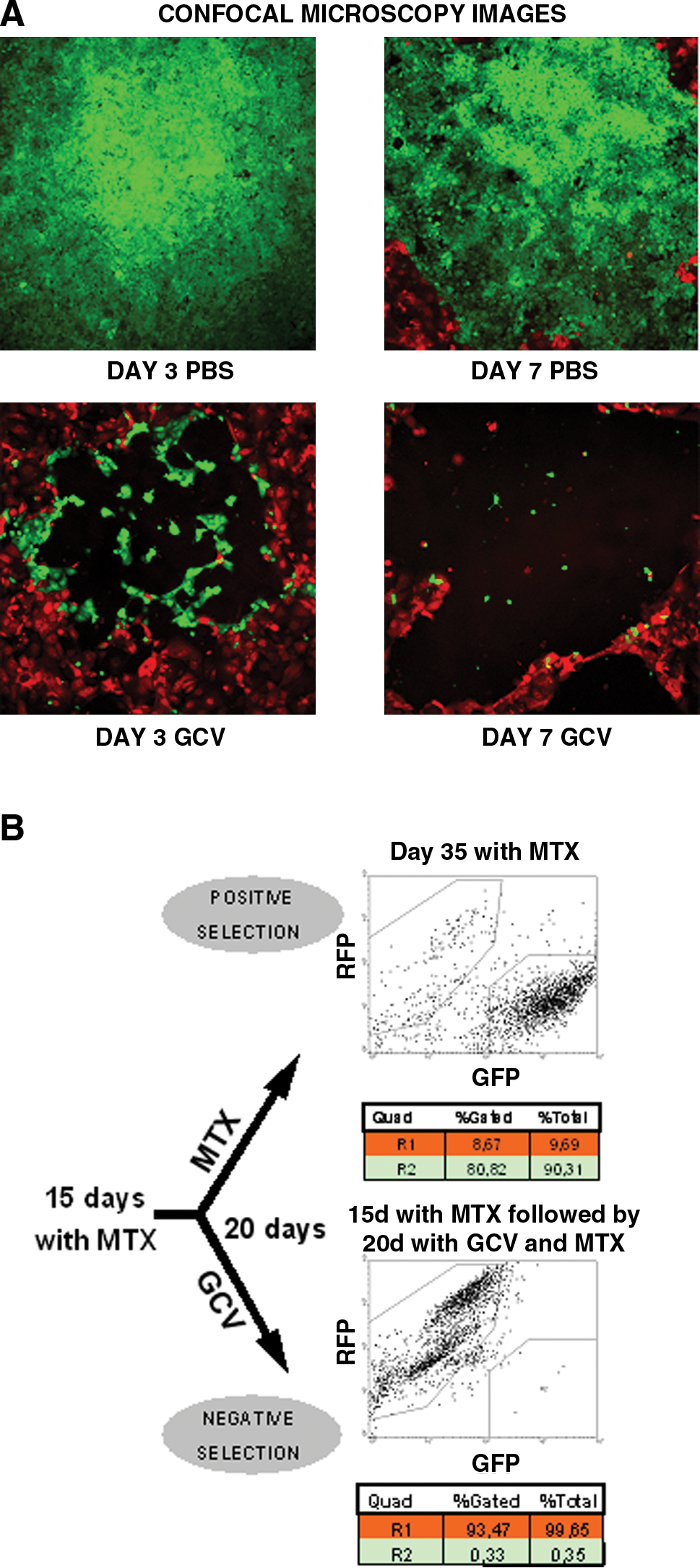
HCT116DHFR/TK-GFP cells (green) are eliminated in vitro with GCV. (
In vivo positive selection with MTX
We determined whether a small proportion of HCT116DHFR/TK-GFP cells could replace parental HCT116 cells in vivo under MTX treatment. In preliminary experiments, mice with HCT116 implanted tumors treated with MTX showed only partial reduction of tumor size (15% on day 17). This suggested that MTX treatment was less efficient in vivo than in vitro (data not shown). We decided to use a higher transfected cell proportion for in vivo positive selection (1%) than for in vitro selection (0.1%). Thus we established subcutaneous tumors in mice by injecting them with a mixture of transfected and nontransfected cells. Animals were given MTX or PBS and were killed on various days throughout the treatment. Regarding MTX toxicity, weight loss did not exceed 10% of body weight, no mice died, and there were no gross signs of toxicity. To measure the relative amounts of parental and transfected cells in tumors treated with MTX, tumor sections were analyzed by fluorescence microscopy. GFP+ transfected cells distributed heterogeneously, and different microscopy fields throughout the sections (×100 and ×400) were used to estimate the percentage of GFP and RFP cells. The proportion of transfected tumor cells increased on MTX treatment. Figure 5A shows a classification of tumors in three groups according to the estimated proportion of transfected cells. Without MTX (Fig. 5A, PBS, open columns) all tumors had <5% of transfected cells on day 31 of treatment. In contrast, on MTX selection (Fig. 5A, MTX, solid columns) more than 40% of tumors had >15% transfected cells at this date. Figure 5B shows three images (×400) of a representative tumor from the MTX group compared with one from the PBS group. A clear enrichment of GFP+ transfected cells is observed.
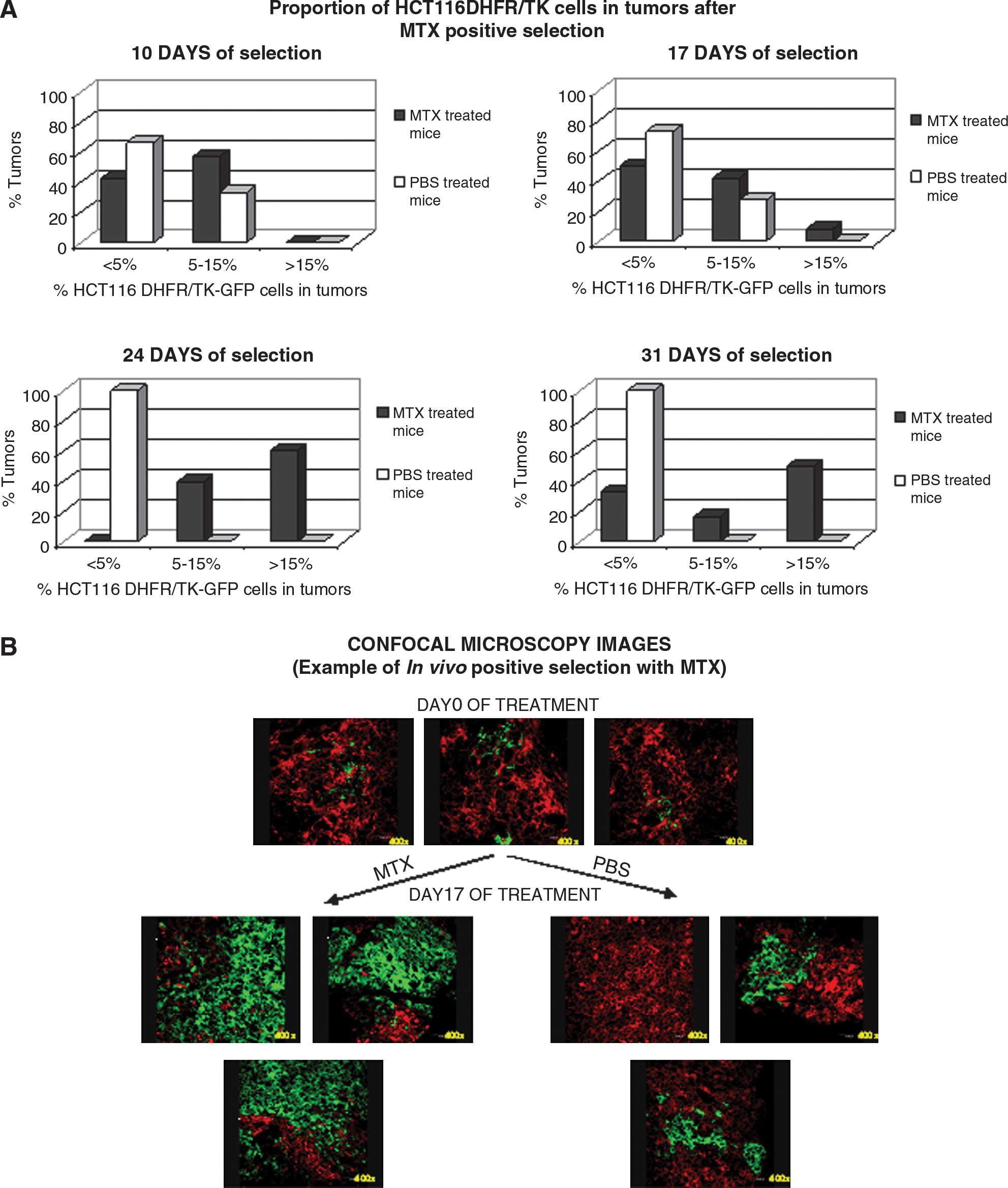
In vivo positive selection of HCT116DHFR/TK-GFP cells. Subcutaneous xenografts were established in nude mice by inoculation of a mixture of 3.5 × 106 cells containing 1% HCT116DHFR/TK-GFP cells and 99% HCT116-RFP cells. Fifteen days later animals were treated with MTX (9 mg/kg; solid columns) or PBS (open columns) intraperitoneally three times per week. On the indicated day of selection, animals were killed and tumors were processed for confocal microscopy. To compensate for cell heterogeneity within tumors, three images (×100 and ×400) were taken from the same section. (
In vivo bystander effect of HCT116DHFR/TK-GFP cells with GCV
To assess whether HCT116DHFRTK-GFP cells induced a bystander effect in vivo, mice with implanted tumors containing 100% HCT116-RFP cells, 100% HCT116DHFR/TK-GFP cells, or a mixture of 50% of each were treated with GCV. Regarding GCV toxicity, weight loss did not exceed 10% of body weight, no mice died, and there were no gross signs of toxicity. This experiment showed that after GCV treatment tumors containing 100% genetically modified cells were eliminated, whereas tumors with parental cells or with only 50% modified cells continued growing (Fig. 6). These results confirmed that although HCT116DHFRTK-GFP cells were sensitive to GCV in vivo, tumors with 50% modified cells were not eliminated because of the absence of a bystander effect in this tumor model.

In vivo bystander effect of HCT116DHFR/TK-GFP cells. Subcutaneous xenografts were established in nude mice by inoculation of a mixture of 3.5 × 106 cells containing 100% HCT116-RFP cells, 100% HCT116DHFR/TK-GFP cells, or a mixture of 50% of each cell type. Once tumors were palpable, animals started GCV treatment. Tumor growth relative to the first day of GCV treatment is shown (n = 6–8). * p < 0.05, significant difference between the HCT116 group and the HCT116DHFR/TK-GFP group; # p < 0.05, significant difference between the MIX 50% HCT116DHFR/TK-GFP group and the HCT116DHFR/TK-GFP group.
In vivo negative selection with GCV
Despite the absence of a bystander effect in this tumor model we determined whether the positive and negative selection strategy was effective in vivo. When tumors containing 1% HCT116DHFR/TK-GFP cells, under positive selection (MTX treatment), reached 300 mm3 we started the negative selection phase (GCV treatment). The mean growth of tumors treated with GCV after positive selection was 50% lower than that of the control group on day 14. However, growth differences were not statistically significant (Fig. 7A). When sections of the tumors were observed under fluorescence, no GFP+ cells were present throughout the tumors of GCV-treated mice (data not shown). This meant that, starting with an initial proportion of 1% HCT116DHFR/TK-GFP cells, although positive selection increased the proportion of transfected cells in tumors (as demonstrated in Fig. 5) and the transfected cells were sensitive to GCV, the proportion of transfected to nontransfected cells was not high enough to abrogate tumor growth on GCV treatment.

In vivo negative selection with GCV. (
When the initial proportion of HCT116DHFR/TK-GFP cells was 6%, negative selection was initiated when tumors were palpable, and animals received a higher dose of GCV (75 mg/kg daily for 14 days). Tumors treated with GCV (negative selection) but without positive selection with MTX did not show any difference from control groups, indicating that 6% HCT116DHFR/TK-GFP cells in a tumor was not enough to obtain a therapeutic effect with GCV. However, the mean growth of tumors treated first with MTX (positive selection) and later with GCV (negative selection) showed statistically significant differences from all control groups (Fig. 7B). This indicates that the positive selection phase was required to increase the initial proportion of modified cells and to decrease tumor growth after GCV treatment.
Discussion
Cancer gene therapy efficacy depends on transducing a high proportion of tumor cells in vivo. To improve gene transfer, modified viral vectors have been used but still the proportion of transfected tumor cells is limited (Rainov, 2000; Rainov and Ren, 2003; Immonen et al., 2004). This limitation may be addressed in vivo by providing a selective advantage to transfected tumor cells (positive selection). After increasing DHFR/TK-transfected cells during a course of positive selection with MTX, the tumor may become sensitive to GCV and eradicated by GCV treatment. The bystander effect could eliminate tumors even with incomplete replacement of tumor cells (Freeman et al., 1993). The transduction of chemoresistance and chemosensitivity genes as a fusion protein in an episomal plasmid increases the safety of this strategy because it reduces the possibility that only the chemoresistance gene is expressed in the target cells.
Despite obtaining an effective level of positive–negative selection in vitro, we could not eradicate all tumor cells in vivo. Several factors can explain these results. First, although DHFR/TK-transfected tumor cells increased proportionally in the tumors, in vivo positive selection with MTX did not result in population replacement as efficiently as in vitro. Whereas the in vitro experiments showed that the transfected population could increase from 0.1 to 90% (Fig. 3B), in vivo only 15% transfected cells could be reached from the 1% initial proportion (Fig. 5A). Poor bioavailability of MTX in vivo could explain these results. We tried various MTX administration schedules and ultimately chose a schedule of 9-mg/kg MTX three times per week because there was a 15% decrease in HCT116 tumor growth on day 17 of treatment compared with mock-treated mice (data not shown). More aggressive schedules resulted in MTX-limiting toxicity. Second, the fast growth rate of HCT116 cells did not allow a long period of positive and negative selection. Third, when we generated tumors with 1% transfected tumor cells and then began MTX treatment, transfected cells formed individual colonies. Thus the increase in transfected tumor cells was heterogeneous in the tumors and the bystander effect would not be as efficient as in transfected cells homogeneously distributed throughout the tumors. Moreover, we observed great heterogeneity in the proportion of transfected cells among MTX-treated tumors, indicating that under the same conditions cell competition was highly variable. When we used an initial transfected population of 6% an increment of transfected cells after positive selection was obtained and their distribution was more homogeneous in the tumors and among MTX-treated tumors. In this way we achieved a proportion of transfected cells to nontransfected cells that was sufficient to decrease tumor growth in treated mice in a model without any bystander effect. Fourth, although HCT116DHFR-TK-GFP cells are sensitive to GCV in vitro (Fig. 2B) and in vivo (Fig. 6), this model did not show any bystander effect. In vitro this lack of bystander effect is clear from the presence of a parental cell monolayer that remains intact around the plaques left by the dying colonies of transfected cells (Fig. 4A). In a bystander effect experiment mixing HCT116DHFR/TK-GFP cells with HCT116-RFP cells at various proportions, we confirmed in vitro that transfected cells could not induce the death of nontransfected cells on GCV treatment (data not shown). Moreover, tumors containing 50% HCT116DHFR/TK-GFP cells plus 50% HCT116-RFP cells continued growing after GCV treatment, confirming the absence of any bystander effect in this tumor model in vivo. Therefore, although we had effective positive selection, and transfected cells were indeed sensitive to the prodrug, a bystander effect would be required to eliminate all the tumors. In any case, it is also important to note the statistically significant therapeutic effect obtained with a positive and negative selection strategy in a model without any bystander effect (Fig. 7B).
We are now applying this positive–negative selection system to a tumor model that has a higher bystander effect of HSV-TK/GCV therapy because of high content of gap junctions (Carrio et al., 2001). In this model the observed increase from 1% to more than 15% TK-positive cells that occurred during the positive selection period could be enough to eradicate all tumors. Burrows and colleagues (2002) presented the TK bystander effect as a percentage of dye transfer in eight glioma cell lines (mean, 65.3; SE, 8.0), seven melanoma cell lines (mean, 45.8; SE, 9.3), and nine colorectal carcinoma cell lines (mean, 12.2; SE, 7.1) including HCT116. Dye transfer-quantified gap junction intercellular communication is implicated directly in the efficacy of the bystander effect (Nicholas et al., 2003). HCT116 had a low level of gap junctions, in accordance with the lack of bystander effect we have observed. To further increase the bystander effect of TK we are also exploring the use of TK modified with the HIV Tat protein transduction domain (Cascante et al., 2005).
Alternatively, other genes for positive–negative in vivo selection may be used. Unger and colleagues have reported a similar strategy to enrich tumor cells before suicide gene therapy (Unger et al., 2007). A fusion gene of cytosine deaminase and uracil phosphoribosyltransferase (FCU1 fusion gene) was used along with N-(phosphonacetyl)-
In general, the proper selection of candidate genes for positive and negative selection must take into account several parameters. For safety, the gene used for positive selection should not confer resistance to a drug used in the clinical management of the tumor to be treated. For example, as MTX may be used in lymphoma treatment, DHFR should not be used for positive selection in this tumor type because it would confer resistance to MTX if the gene therapy was given previous to or concomitant with the chemotherapy. If the competition gene therapy were applied after MTX-based chemotherapy, relapsed tumors would already be resistant to MTX and there would not be any possibility for positive selection. Although a fusion gene such as DHFR/TK may be used in an effort to minimize the possibility of conferring positive selection without the accompanying suicide mechanism, one cannot ensure that the TK gene becomes nonfunctional by mutation, and then MTX resistance would pose a problem to the established clinical treatment. For these reasons MTX positive selection may be an option for tumors other than lymphomas. On the other hand, competition gene therapy must confer resistance to a drug that has a negative effect in the nontransduced part of the tumor, although such a drug is not used in the clinical treatment. It is possible to choose genes that confer resistance to drugs used as third or later lines of treatment. In colon carcinoma, it would not be appropriate to confer resistance to 5-FU, oxaliplatin, leucovorin, irinotecan, capecitabine, and mitomycin C, but it might be considered acceptable to use aldehyde reductase or alkylguanine DNA-transferase to provide resistance to alkylating agents such as cyclophosphamide. Although suicide genes such as TK can induce a bystander effect in models with gap junctions, selecting genes for negative selection that do not depend on gap junctions should improve the prospects of the positive–negative selection strategy.
We have focused on the process of positive–negative selection after establishing tumors with 1 or 6% transfected cells. A pending question for this treatment concerns how the selecting genes are going to be delivered to tumors. Transfection needs to be stable, either by genomic integration or episomal replication and segregation to daughter cells. The pEPI-DHFR/TK plasmid presented here contains these later elements and it could be used in combination with nonviral vectors such as tumor-targeted liposomes to test the competition–suicide strategy after systemic delivery. For a higher transduction rate several viral vectors could be used such as adeno-associated virus (AAV) (Hacker et al., 2005) or helper-dependent adenoviral vectors containing Epstein–Barr virus (EBV) episomal replication elements (Dorigo et al., 2004; Kreppel and Kochanek, 2004) or adeno-transposons (Yant et al., 2002).
In conclusion, we prove the concept that a fusion gene, which confers a selective advantage and a suicide mechanism, can be used for competition–suicide cancer gene therapy in which gene-modified cells outcompete tumor cells (positive selection) and are later eliminated (negative selection).
New drug resistance genes should be tested to improve positive selection in vivo and new prodrug-converting genes should be tested to improve the bystander effect and negative selection in vivo.
Footnotes
Acknowledgments
The authors thank Neus Bayo-Puxan, Alena Gros, Juanjo Rojas-Exposito, Sonia Guedan, and Marta Gimenez for technical assistance. The authors thank Esther Castaño for technical support in flow cytometric data analysis and Benjamin Torrejon for technical support in the use of confocal microscopy. J.M-Q. was supported by a graduate student fellowship from the Fundació Privada Institut d'Investigació Biomèdica de Bellvitge (IDIBELL). Funding was provided by BIO2005-08682-C03 from the Ministerio de Educación y Ciencia, by the Departament d'Universitats, Recerca i Societat de la Informació from the Generalitat de Catalunya (2005 SGR 00727), and by the CIBER de Enfermedades Raras, an initiative of the ISCIII. R.A. belongs to the Network of Cooperative Research on Cancer (C03-10), Instituto de Salud Carlos III of the Ministerio de Sanidad y Consumo, Government of Spain.
Author Disclosure Statement
The authors declare no competing financial interests.