
Abstract
Duchenne muscular dystrophy (DMD) is a myodegenerative disorder caused primarily by mutations that create premature termination of dystrophin translation. The antisense oligonucleotide approach for skipping dystrophin exons allows restoration of the correct reading frame in the dystrophin transcript, thus producing a shorter protein. A similar approach in humans would result in the conversion of DMD to the milder Becker muscular dystrophy. It has been demonstrated previously that repeated intravascular injection of phosphorodiamidate morpholino oligomers (PMOs) in the mdx mouse induces more dystrophin expression than a single injection, but this approach is costly, and data demonstrating the safety of high doses of systemically injected PMO are unavailable. Furthermore, several publications have demonstrated the efficacy of peptide-conjugated PMOs, but the clinical applicability of such compounds is unclear at this stage. Here, we report that multiple intravascular injections of low doses of naked PMO show significantly more dystrophin-positive fibers in a variety of muscle groups, 8 weeks after administration compared with a single dose of the same total amount. After administration of a total of 200 mg of PMO per kilogram, histological features, such as the cross-sectional area, centronucleation index, and expression of the dystrophin-associated protein complex, showed significant improvement in mice treated by repeated injection. Furthermore, four administrations of just 5 mg/kg induced a significant amount of dystrophin expression. These results clearly demonstrate the key role of the optimization of dosing regimen for the systemic administration of PMO in patients, and support the clinical feasibility of this approach with naked PMO.
Introduction
Patients with DMD and animal models of the disease exhibit a small number of so-called revertant muscle fibers capable of expressing dystrophin. These arise by the exclusion, during the process of pre-mRNA splicing, of either the exon carrying the mutation, or of additional dystrophin exons that are unaffected by the existing genomic deletion (Sherratt et al., 1993; Lu et al., 2000). This mechanism has been exploited by the use of an antisense oligonucleotide (AO) strategy, in which the deliberate and targeted skipping of one or more dystrophin exons increases the efficiency of the restoration of the correct reading frame in the dystrophin transcript (Wilton et al., 1997; Dunckley et al., 1998; Graham et al., 2004; Lu et al., 2005; Alter et al., 2006; van Deutekom et al., 2007). If applied to humans, this method would allow the conversion of the DMD phenotype to the milder Becker muscular dystrophy (Monaco and Kunkel, 1988).
Several parameters must be considered to increase the efficacy of this approach, such as finding the best performing AO, the route of delivery, and the optimal dosing regimen. Various chemistries have been used for both in vitro and in vivo studies, including DNA, phosphorothioate-linked 2′-O-methyl RNA (2OMePS), locked nucleic acid (LNA), 2′-O,4′-C-ethylene-bridged nucleic acid (ENA), peptide nucleic acid (PNA), and phosphorodiamidate morpholino oligomer (PMO) (Dunckley et al., 1998; Gebski et al., 2003; Aartsma-Rus et al., 2004; Surono et al., 2004; Lu et al., 2005; Alter et al., 2006; Takeshima et al., 2006; Yin et al., 2008a).
Two independent ongoing clinical trials in the Netherlands and the United Kingdom are using 2OMePS and PMO, respectively (van Deutekom et al., 2007; Muntoni et al., 2008). Some reports have shown that PMO appears superior to 2OMePS, at least for in vivo applications, thanks to the higher affinity for the sequence target and the better resistance to endonucleases (Gebski et al., 2003; Fletcher et al., 2006; Amantana et al., 2007). Moreover, PMO can easily be modified to link effective cell-penetrating peptides to improve the uptake by target tissues (Fletcher et al., 2006; Amantana et al., 2007; Jearawiriyapaisarn et al., 2008; Wu et al., 2008; Yin et al., 2008b). The efficacy of AO-derived exon skipping was first demonstrated using in vitro cultures of murine and human myoblasts (Dunckley et al., 1998; Aartsma-Rus et al., 2003) and then in vivo after intramuscular injection, where this approach allowed skipping of the mutated exon 23 in the mdx mouse (Gebski et al., 2003; Lu et al., 2003). More recently, dystrophin expression in a wide variety of muscle groups was achieved in mdx mice using systemic approaches such as intravenous, intraperitoneal, and intraarterial administrations of AO (Lu et al., 2005; Alter et al., 2006; Fletcher et al., 2006; Adams et al., 2007; Vitiello et al., 2008). Moreover, life-long expression of dystrophin produced by exon skipping was achieved by delivery of adeno-associated viral (AAV) vectors expressing U1 or U7 small nuclear RNA (snRNA) genes containing antisense sequences (Goyenvalle et al., 2004; Denti et al., 2006, 2008), or by the administration of stem cells transduced with those constructs (Benchaouir et al., 2007; Meregalli et al., 2008).
The systemic administration of PMO presents some difficulties due largely to its accumulation in the kidney, lung, and liver in the hours after administration (Amantana and Iversen, 2005). Some reports have highlighted the opportunity to inject AOs repeatedly to induce higher levels of dystrophin expression, due to a cumulative effect of administered AO (Alter et al., 2006; Fletcher et al., 2006). This approach is costly and data that demonstrate the safety of high dose of systemically injected PMO are not available. For these reasons, the search for better methods for reducing the quantity of AO, while at the same time improving the efficacy of dystrophin expression, is crucial. Here, we show that after systemic intravenous administration of naked PMO, the expression of dystrophin is kept high with no apparent reduction for at least 8 weeks, confirming that the PMO may require fewer administrations than other AOs used in the systemic treatment of DMD. More importantly, we report that multiple systemic injections of a lower amount of PMO show significantly more dystrophin-positive fibers in a wide variety of muscle groups than after administration of a single dose with the same total amount, 8 weeks after administration. In particular, the lower dosage we tested represents a clinically applicable amount for a systemic treatment in humans. These findings highlight that the dosing regimen of PMO administration is a fundamental parameter to be considered to reduce the quantity of naked PMO necessary for systemic delivery.
Materials and Methods
In vivo injection of antisense oligonucleotide reagents
Phosphorodiamidate morpholino oligomer (GGC CAA ACC TCG GCT TAC CTG AAA T) (Gebski et al., 2003), designed to skip exon 23 of dystrophin in the mdx mouse, was purchased from Gene Tools (Philomath, OR), and was diluted in sterile saline (Sigma-Aldrich, Poole, UK) to give a total volume of 30 μl for intramuscular injections and 200 μl for intravenous injections. All the C57BL/10ScSn-Dmdmdx (mdx) mice used in this study were 6 months old at the start of the experiments. For intramuscular injection, mdx mice were anesthetized via intraperitoneal injection of 3 μl/g body weight of 25% (v/v) fentanyl–fluanisone and 25% (v/v) midazolam in sterile water and AOs were injected into the left tibialis anterior (TA) muscle while the contralateral muscle was sham injected with saline. Intravenous injection of PMO was performed via the tail vein, without anesthesia, after warming of the mice in a 40°C incubator for 10 min. For all the experiments involving systemic administration, untreated age- and sex-matched animals were used as negative controls. Animals were monitored hourly for 4 hr and then daily for the remainder of the experiment. At the indicated time points, muscles were harvested, mounted in O.C.T. compound (Raymond A Lamb/Thermo Fisher Scientific, Eastbourne, UK), frozen in 2-methylbutane (isopentane) chilled with liquid nitrogen, and stored at −80°C. The muscles harvested were TA for the intramuscular injection experiments; TA, extensor digitorum longus (EDL), soleus, diaphragm, and heart for the experiment examining the persistence of dystrophin expression after systemic delivery; TA, EDL, soleus, quadriceps, gastrocnemius, diaphragm, triceps brachii, and heart for the dose regimen experiment.
Animals were bred in-house and food and water were provided ad libitum. They were maintained, and in vivo experimentation was conducted, under statutory Home Office recommendation, regulatory, ethical, and licensing procedures, and under the Animals (Scientific Procedures) Act 1986 (project license PPL 70/6160).
Muscle sectioning and immunohistochemistry
Mounted muscles were sectioned at a thickness of 10 μm, using a Bright OTF 5000 cryostat (Bright Instruments, Huntingdon, UK). The tissue sections were placed on coated glass slides (VWR, Lutterworth, UK) and stored at −80°C before use. The following antibodies were used for immunohistochemical staining: NCL-DYS2 (monoclonal, anti-dystrophin; diluted 1:50), NCL-α-SG (monoclonal, anti-α-sarcoglycan; diluted 1:50), NCL-β-DG (monoclonal, anti-β-dystroglycan; diluted 1:50) (all from Novocastra Laboratories, Newcastle, UK), P6 (rabbit polyclonal, anti-dystrophin; diluted 1:400) (Sherratt et al., 1992), anti-laminin (rat monoclonal; diluted 1:1000; Sigma-Aldrich), and anti-NOS1 (nitric oxide synthase-1, rabbit polyclonal; diluted 1:50; Santa Cruz Biotechnology, Santa Cruz, CA). For staining of mouse muscle sections with mouse monoclonal antibodies, a Vector M.O.M. fluorescein immunodetection kit (Vector Laboratories, Peterborough, UK) was used also in conjunction with streptavidin–Alexa Fluor 568 (Invitrogen, Paisley, UK) for the staining of dystrophin, and where necessary for the double staining of dystrophin-associated protein complex (DAPC) components. All staining was carried out at room temperature under humidified conditions and prepared according to the M.O.M. manufacturer's instructions. Quantification of dystrophin was performed on the section with the highest number of positive fibers from a set of sections that covered the entire length of muscle. Dystrophin-positive fibers were manually counted with SigmaScan Pro image analysis software (Systat Software, Hounslow, UK). The dystrophin staining was also used for analysis of the cross-sectional area of dystrophin-positive fibers. The percentage of fibers expressing DAPC was calculated by counting the number of double-positive fibers for dystrophin and DAPC components in five randomly captured fields for every muscle (magnification, × 20). A section representative of the total muscle was chosen. Where the counting of fibers was performed, all fibers that showed even moderate staining for a marker were considered positive. The images were recorded and processed using identical parameters of exposure, saturation, and gamma between differentially treated and untreated specimens.
Histological analyses
A standard protocol was used for hematoxylin and eosin (H&E) staining. This staining was used to calculate the average number of fibers in the muscles of 6-month-old mice (TA, 3300 ± 244; quadriceps, 8465 ± 627; diaphragm, 4409 ± 624; gastrocnemius, 6381 ± 2026; EDL, 968 ± 103; soleus, 683 ± 467; n = 3) and to verify the percentage of centrally nucleated fibers. The Vector M.O.M. immunoperoxidase kit was used with a 3,3′-diaminobenzidine (DAB) substrate kit (Vector Laboratories) for the staining with laminin antibody (in accordance with the M.O.M. manufacturer's instructions) in TA, quadriceps, and diaphragm in order to analyze the cross-sectional area (CSA) of fibers. Pictures of 6 random fields for TA and diaphragm and 12 for quadriceps were taken. Determination of the total fiber number in muscles and the analysis of CSA of total muscle fibers were performed with SigmaScan Pro image analysis software.
Western blot analyses
Protein concentration was measured using a RC DC protein assay (Bio-Rad, Hemel Hempstead, UK). Samples were diluted in loading buffer (100 mM dithiothreitol [DTT], 75 mM Tris-HCl [pH 6.8], 10% sodium dodecyl sulfate [SDS], 20% glycerol) and run for 1 hr on 3–8% Tris–acetate SDS–polyacrylamide gels (Invitrogen). The same quantity of protein for each sample was run on the gel and then transferred to nitrocellulose (Hybond; GE Healthcare, Little Chalfont, UK) for 1.5 hr at 30 V. Nonspecific binding was prevented by blocking with 5% dried milk powder in PBST for 2 hr at room temperature. α-Tubulin expression was evaluated with a mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich) and dystrophin expression was evaluated with the mouse monoclonal antibody MANEX 1011 (Lu et al., 2000), recognizing an epitope in exons 10 and 11 in the N-terminal domain, diluted 1:100 in phosphate-buffered saline–Tween (PBST) overnight at 4°C followed by horseradish peroxidase (HRP)-conjugated anti-mouse (Dako, Ely, UK) diluted 1:1000 in PBST for 1 hr at room temperature. The membrane was finally placed in enhanced chemiluminescence (ECL) solution for 1 min and exposed to film (Hyperfilm-ECL; GE Healthcare) for up to 5 min depending on the loading of the samples. Densitometric analysis of bands was performed with ImageJ 1.4 software (National Institutes of Health, Bethesda, MD).
Statistical analysis
Data are expressed as means ± standard error of the mean. The Student two-tailed t test was used for all analyses. Values of p less than 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001). For all statistical analyses, the GraphPad Prism software package (version 4; GraphPad Software, San Diego, CA) was used.
Results
De novo dystrophin expressed after treatment with phosphorodiamidate morpholino oligomer persists for at least 2 months
We first verified the persistence of de novo dystrophin expression after a single intravenous administration of PMO. PMO was administered via the tail vein at doses of 1, 10, and 100 mg/kg, and animals were killed 4 or 8 weeks after injection. Figure 1 shows the number of dystrophin-positive fibers found after immunostaining of tibialis anterior (TA), diaphragm, extensor digitorum longus (EDL), and soleus muscles. At the lowest dose, the number of dystrophin-positive fibers in all muscles analyzed at week 4 and week 8 was barely above the level of revertant fibers observed in untreated mdx mouse muscles (from a maximal number of 8 fibers in soleus to 26 fibers in TA). The highest dose of PMO gave rise to the highest number of dystrophin-positive fibers in all the muscles tested, showing a dose–response correlation between dose of AO and dystrophin expression (Fig. 1). More importantly, no significant difference was observed between the average number of dystrophin-positive fibers at the two time points analyzed, indicating that fibers expressing dystrophin persist for at least 8 weeks after intravenous delivery of PMO. No dystrophin-positive cardiomyocytes were observed in cardiac muscle of treated or untreated mice (data not shown).
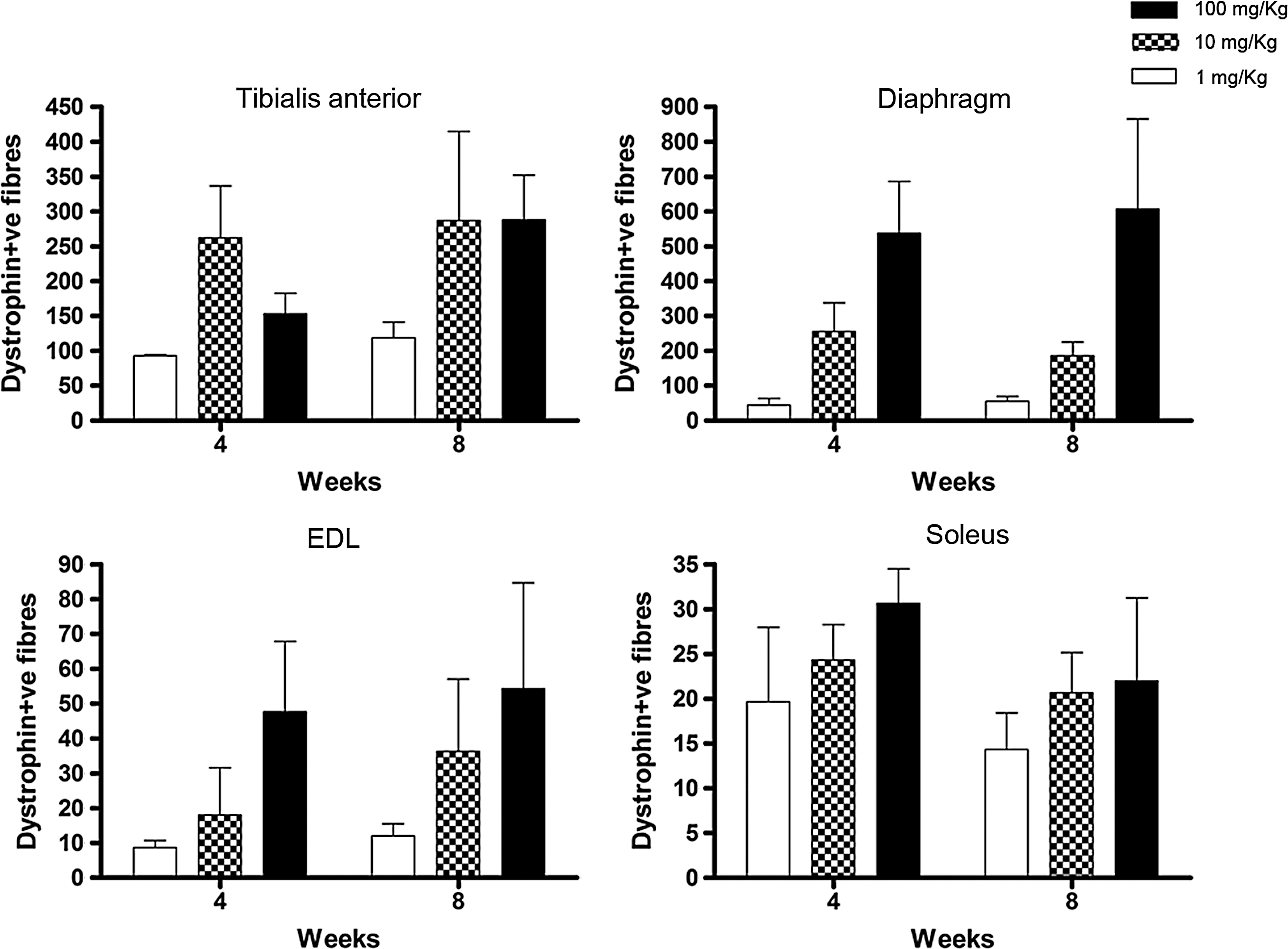
Persistence of de novo dystrophin expression after intravenous injection of phosphorodiamidate morpholino oligomer (PMO). Graphs show the number of dystrophin-positive fibers detected by immunostaining of tibialis anterior (TA), diaphragm, extensor digitorum longus (EDL), and soleus muscles, 4 and 8 weeks after administration of PMO at 1, 10, or 100 mg/kg, via the tail vein. Quantification of dystrophin-positive fibers was performed on the section with the highest number of positive fibers from a set of sections that covered the entire length of muscle. Data are expressed as the mean number (and SEM) of dystrophin-positive fibers per muscle (n = 3). The difference in dystrophin expression 4 and 8 weeks after any treatment was not statistically significant by two-tailed Student t test (n = 3).
The sustained expression of dystrophin in response to PMO administration was confirmed by intramuscular injection of 3 nmol (20 μg) of PMO into TA muscles of 6-month-old mdx mice. This demonstrated widespread dystrophin expression up to week 10 postinjection, covering approximately 30% of the total section area (data not shown).
Dividing a single large dose of PMO into four equal doses improves dystrophin expression in skeletal muscle
It has been reported previously that after several injections of the same quantity of AO, the cumulative effect on dystrophin expression greatly increases the number of dystrophin-positive fibers (Lu et al., 2005; Alter et al., 2006). To clarify the role of repeated administrations in increasing the dystrophin expression in mdx muscles, animals were injected intravenously with 200 mg/kg (group A) or weekly four times with 50 mg/kg (group B). Because we had previously verified that dystrophin expression persists for at least 2 months after PMO delivery, animals were killed 8 weeks after the first injection. After these administrations, the multiple injection regimen yielded the highest number of dystrophin-positive fibers in all the muscles analyzed, with the best result for quadriceps, in which the number of fibers was increased 7-fold, from 8% in group A to 57% in group B (p = 0.0005) (Fig. 2a). In diaphragm (p = 0.026), gastrocnemius (p = 0.0016), and TA (p = 0.007), the number of dystrophin-positive fibers increased by approximately 4-fold, whereas EDL and soleus showed a generally lower percentage of dystrophin-positive fibers for both groups A and B, with differences that were not statistically significant.

Effect of dosing regimen on the level of dystrophin expression after high-dose administration of PMO. (
Examples of the general appearance and distribution of dystrophin-positive fibers are shown for muscles from untreated mdx (Fig. 2b), treatment group A (Fig. 2c), and treatment group B (Fig. 2d). Interestingly, most of the dystrophin-positive fibers in quadriceps were localized in the vastum lateralis and medialis, with fewer patches of dystrophin-positive fibers present in the rectus femoris.
Western blotting analysis shows that the amount of de novo dystrophin protein expressed by treated muscles of group B increased 3-fold in TA, more than 2-fold in quadriceps, and 2-fold in diaphragm, compared with muscles of group A (Fig. 3 and data not shown). When compared with C57BL/10 (wild-type) mouse muscle, Western blots showed up to 15–17% dystrophin expression for quadriceps and diaphragm and up to 10–13% for TA. Importantly, whereas for TA and quadriceps the amount of dystrophin was similar in the three treated samples, only one diaphragm of the three showed evidence of dystrophin expression by Western blot (data not shown). No dystrophin-positive cardiomyocytes were observed in either treated or untreated heart (data not shown).

Dystrophin expression 2 months after intravenous administration of a high dose of PMO. Top: Forty micrograms of total protein extracted from quadriceps and TA muscles of each of the mice from the three treatment groups, and 10 μg of protein extracted from C57BL/10 mouse tibialis anterior muscle (WT TA), were analyzed by Western blot analysis for dystrophin (dys) and α-tubulin (α-tub). Bottom: Densitometric analysis of bands from the immunoblot for dystrophin and α-tubulin from quadriceps and TA samples. The averages were obtained with the values of dystrophin normalized to the corresponding bands for α-tubulin, followed by comparing these values with the ratio obtained in the same way for a C57BL/10 TA muscle (which was considered as 100%). Data are expressed as means and SEM (n = 3). The final comparison between the two treated groups was performed with a two-tailed t test (*p < 0.05).
Histological analysis of tibialis anterior, quadriceps, and diaphragm shows partial recovery of wild-type morphology and DAPC expression after high-dose administration
To verify that the increase in dystrophin expression was sufficient to rescue, at least partially, the dystrophic morphology of mdx muscle, the percentage of centrally nucleated fibers and the fiber cross-sectional area (CSA) were analyzed. The results for TA, quadriceps, and diaphragm are shown in Fig. 4a. The quadriceps, the muscle showing the highest number of dystrophin-positive fibers, presented a significant difference in centrally nucleated fibers only between treatment group B and the untreated mdx mice. TA and diaphragm muscles of group B did not show a significantly different number of centrally nucleated fibers, compared with the untreated mdx mice. However, after hematoxylin and eosin (H&E) staining, the cross-sectional area (CSA) of muscle fibers in TAs of group B was significantly more homogeneous than those from untreated mdx muscles (Fig. 4b and c). We then compared the distribution of fiber CSA in TA, quadriceps, and diaphragm after treatment with four injections (group B) with that of untreated mdx mice. Whereas quadriceps and diaphragm showed an unaltered distribution of fiber CSA after treatment (χ2 analysis, in quadriceps χ2 = 2.3, df = 14, p > 0.25; in diaphragm χ2 = 5.18, df = 10, p > 0.25; distributions not shown), a significant difference was present for TA (χ2 analysis; χ2 = 60, df = 12, p < 0.0001) (Fig. 4d), in which the group B treated muscles showed a distribution of many more fibers with a larger area. The distributions of CSA for muscles of group A and untreated animals were not significantly different in TA (χ2 = 15.98, df = 12, p > 0.15). Interestingly, the dystrophin-positive fibers in all the muscles analyzed for both the treated groups showed a significant increase in CSA compared with the dystrophin-negative fibers of untreated mdx muscle (Fig. 4e).
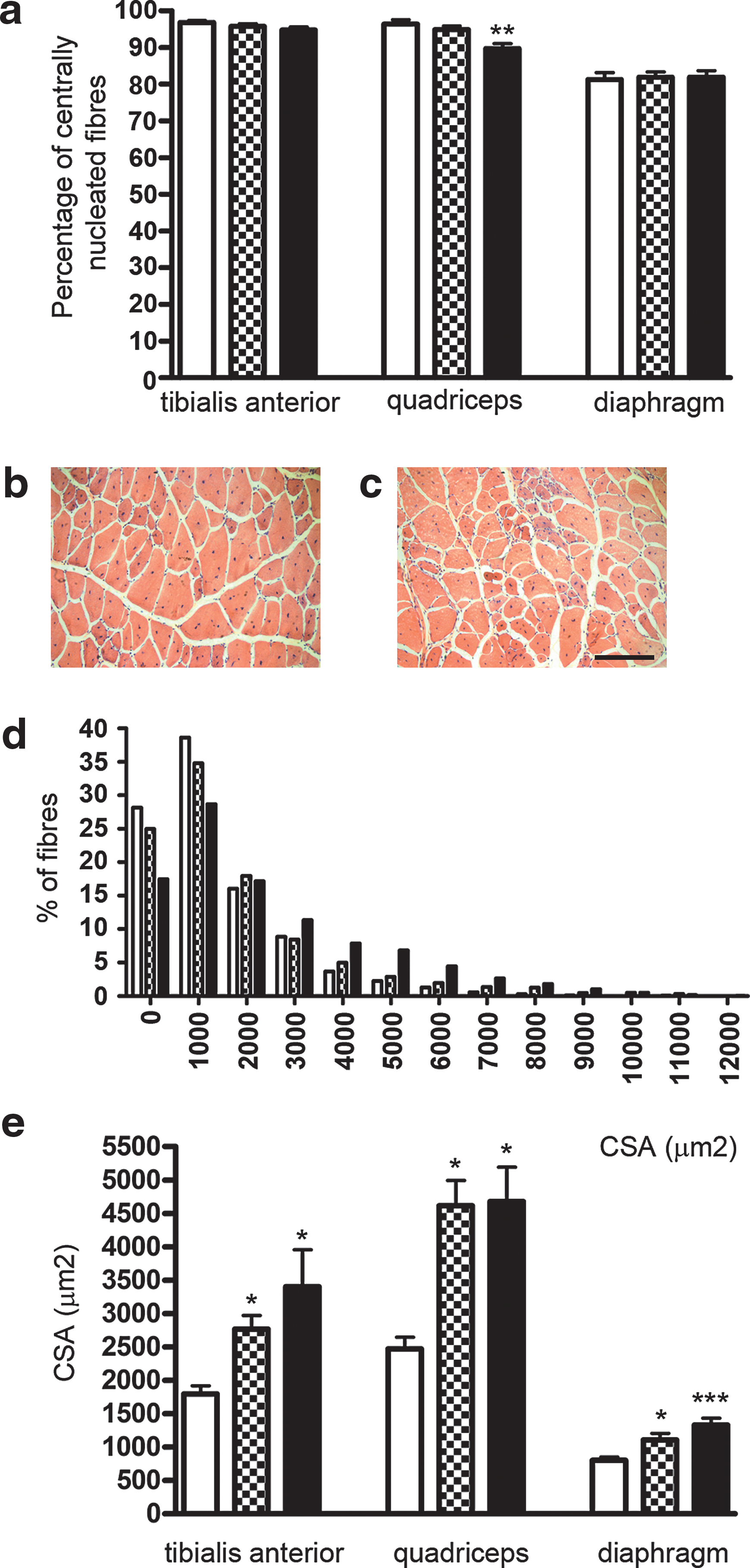
Morphometric analyses of muscles 2 months after PMO administration. Data are shown for TA, quadriceps, and diaphragm muscles from untreated mdx mice (open columns) and mice treated with one injection of PMO at 200 mg/kg (checkered columns) or with four injections of PMO at 50 mg/kg (solid columns). (
Immunofluorescence analysis of serial sections showed the expression of components of the dystrophin-associated protein complex (DAPC) to be directly related to the staining of dystrophin in the muscles analyzed (Fig. 5). The presence of α-sarcoglycan, β-dystroglycan, and neuronal nitric oxide synthase (nNOS, or NOS1) was compared between groups A and B. This is shown graphically (Fig. 5a) and by immunofluorescence, demonstrating the coincidence of muscle fibers positive for all DAPC components tested and dystrophin, as indicated by the asterisks in Fig. 5b. The highest correlation between the presence of DAPC and dystrophin staining was observed for the quadriceps and TA muscles of group B, where it ranged between 80 and 100% for the three DAPC proteins analyzed. The same trend, albeit without a statistically significant difference, was observed for diaphragm, where the staining was generally lower for all the proteins analyzed, as compared with TA and quadriceps (Fig. 5a).

Detection of dystrophin-associated protein complex (DAPC) components after PMO injection. (
Repeated administration of low doses of PMO results in large increases in dystrophin expression in skeletal muscle, but does not improve histological features
To verify the effect of lower, repeated doses, which would be clinically applicable in humans, we reduced the total amount of PMO administered 10-fold, such that mdx mice were injected intravenously with PMO at 20 mg/kg or with four weekly doses of PMO at 5 mg/kg. As previously reported for the high dose, animals were killed 8 weeks after the bolus single injection or 8 weeks after the first of four weekly injections. As shown in Fig. 6, multiple injections of 5 mg/kg resulted in a significant increase in the number of dystrophin-positive fibers in all the muscles analyzed. The highest number of dystrophin-positive fibers (27%) was observed in the TA muscle. In quadriceps (21%), gastrocnemius (21%), and triceps brachii (26%), more than 20% dystrophin-positive fibers were counted, whereas in diaphragm about 15% of the total fibers were positive for dystrophin (Fig. 6a). Dystrophin staining showed a comparable spread but less intense signal than the staining of sections in muscle treated with the high dose (Fig. 6b–d). Western blot analyses showed dystrophin expression in all quadriceps treated with four weekly injections of 5 mg/kg, whereas the same analysis for TA showed no dystrophin expression (see Supplementary Fig. 1 at

Effect of dosing regimen on the level of dystrophin expression after low-dose administration. (
Discussion
The use of antisense oligonucleotides to induce the skipping of one or more exons and restore the expression of dystrophin is one of most promising approaches to treat Duchenne muscular dystrophy (DMD). This approach would allow the restoration of dystrophin in more than 70% of patients with DMD, particularly in those with short deletions or other mutations in nonessential coding domains (Aartsma-Rus et al., 2009). The 2′-O-methyl phosphorothioate antisense oligoribonucleotide (2OMePS) and phosphorodiamidate morpholino oligomer (PMO) are currently the most promising chemistries and they were chosen by two independent consortia to develop clinical trials in the Netherlands (van Deutekom et al., 2007) and in the United Kingdom (Muntoni et al., 2008). PMO is perhaps more promising for in vivo applications, thanks to the higher affinity for the sequence target and the greater resistance to degradation by nucleases. However, an important disadvantage of PMO is its uncharged nature, which reduces the uptake into cells in in vitro studies and, most important, seems to increase the accumulation of PMO in lung, liver, and kidney within 1–20 hr of systemic injection (Amantana and Iversen, 2005). Although several reports have demonstrated the capacity of PMO to restore dystrophin expression efficiently in vivo, a high amount needs to be administered to obtain widespread expression of dystrophin and functional improvement in skeletal muscles (Lu et al., 2005; Alter et al., 2006). Because of the high cost of PMO and the lack of data showing the safety of long-term treatment, the most important challenge is to reduce the amount of the AO delivered, while at the same time retaining sustained dystrophin expression. The coupling of PMO with specific cell-penetrating peptides (Jearawiriyapaisarn et al., 2008; Wu et al., 2008; Yin et al., 2008b) has shown encouraging results, but no long-term experiments have been reported that could rule out any possible toxic or immunogenic effects. In this study, we chose to use the current best performing unconjugated PMO designed for in vivo use in mdx mice (Alter et al., 2006). The PMO-induced dystrophin expression was strictly correlated with the administered dose, and persisted for at least 8 to 10 weeks after intravenous or intramuscular injection in adult mice. The long half-life of this shorter but functional protein is relevant, because a sustained effect of delivered AO is necessary in order to guarantee as long a time as possible between administrations in a clinical approach. However, the stability of the de novo dystrophin produced will have to be verified directly in patients' cells because, as a result of the exon-skipping mechanism, the variability of different dystrophins produced after the skipping of different exons could result in the expression of dystrophins with different half-lives.
Because treatment with AO modulates pre-mRNA splicing without any alteration at the DNA level, repeat administration is de rigueur. We chose to repeat the injections weekly because that would appear to represent the most feasible clinical regimen of administration. Our results demonstrate that it is possible to increase the number of dystrophin-positive fibers significantly in a wide variety of muscle groups after 8 weeks, simply by increasing the number of administrations, but not the total amount of PMO delivered.
Although we were not able to perform any force-related physiological tests, the number of dystrophin-positive fibers and the amount of protein expressed in TA, quadriceps, and diaphragm after injecting mice with 1.25 mg of PMO (50 mg/kg) four times are comparable to results obtained by Lu's group after three injections of 2 mg of PMO per mouse (Alter et al., 2006), which showed an improvement in the normalized maximal isometric tetanic force for the treated TA muscles of adult animals. Importantly, the morphology of the muscles changed after the treatment with four injections of 50 mg/kg. In quadriceps, which showed the highest number of dystrophin-positive fibers, the percentage of centrally nucleated fibers is decreased, showing a possible change in the cycle of muscle degeneration and regeneration. The number of centrally nucleated fibers in diaphragm was substantially low, irrespective of treatment group, but not in TA and quadriceps. This is probably due to the different process of degeneration–regeneration that affects this muscle, where fibrotic tissue substitutes muscle tissue more quickly (Stedman et al., 1991). Moreover, there was a degree of variability in dystrophin-positive fibers among diaphragms from animals injected with PMO, which we attribute to the variations in the severity of the diaphragm pathology in these animals, despite their similar ages.
The experiments performed by injecting a low dose of PMO showed the same outcome as the high dose, with a significant increase in dystrophin-positive fibers after four injections of the divided dose. The numbers of fibers expressing dystrophin in TA and diaphragm were similar to those obtained after injection of a high amount of PMO, both for one injection with the total amount and for the four injections of the divided dose. However, dystrophin bands in Western blot analysis of low-dose TA muscles were not detectable, suggesting that whereas the qualitative distribution of dystrophin reexpression is similar in all groups, the quantitative levels may differ. This finding is in keeping with research involving PMO conjugated to cell-penetrating peptides, which suggests that the challenge for the PMO is entry into the fibers, rather than delivery to widespread muscle groups. In quadriceps, the percentage of dystrophin-positive fibers after multiple injections was lower compared with TA muscle treated with the same dose regimen. However, in quadriceps the dystrophin produced was sufficient to be revealed by Western blot analysis, suggesting that the lower-proportion positive fibers in this muscle each express more dystrophin protein than those in treated TA muscles. This could perhaps reflect a different permeability to PMO of these fibers due to the major use of the quadriceps compared with the TA muscle.
A mechanism that could explain the increase in dystrophin expression after repeated injections is the clearance, in the first few hours after injection, by liver, lung, and kidneys, of AO that has not been taken up by skeletal muscle (Amantana and Iversen, 2005). This limiting threshold effect could be overcome by smaller aliquots administered at different times, as we have done here; the result would be a greater exposure to PMO. An alternative, but not mutually exclusive model, which remains untested, is one in which the increase in the number of fibers expressing dystrophin observed after repeated injections indicates widespread distribution of AO within the muscle, perhaps by virtue of using different areas of the capillary bed with each administration; delivery of a single bolus could result in clusters of dystrophin-positive fibers surrounding the more major vessels of the vasculature (F.C.T., unpublished observations; Fletcher et al., 2006).
Finally, the presence of a low level of dystrophin pre-mRNA, especially in mdx mice, in which nonsense-mediated decay is expected to destroy the transcript, could result in it being skipped efficiently either by high-dose (50 mg/kg) or low-dose (5 mg/kg) PMO; the subsequent transcripts appearing after the first injection of PMO might then be available for the following administration of PMO. Importantly, the fact that the dystrophin expressed after weekly injection of 5 mg/kg can give rise to comparable numbers of dystrophin-positive muscle fibers as administration of 10 times as much PMO, suggests strongly that there is a relatively low threshold for the amount of PMO administered, and that a detectable and sustained level of dystrophin expression can be achieved after a few injections.
Complications in cardiac muscle are one of the main causes of death in patients with DMD. No data have been published thus far that report an increase in dystrophin expression in cardiomyocytes after delivery of uncoupled 2OMePS (Lu et al., 2005) or PMO (Alter et al., 2006). The same result was observed in our experiments with all of the dosing regimens of PMO tested. However, the use of arginine, 6-aminohexanoic acid, and/or β-alanine improved the uptake of PMO by cardiomyocyte membranes (Jearawiriyapaisarn et al., 2008; Wu et al., 2008; Yin et al., 2008b), demonstrating the feasibility of inducing dystrophin by the exon-skipping approach in cardiac muscle.
In conclusion, we have demonstrated that the choice of dosing regimen is key to the efficiency of dystrophin production by exon skipping in response to PMO in a murine model of DMD, and is likely to be the case in human subjects also. Weekly administration of either high or low levels of naked PMO gave a far superior number of dystrophin-positive fibers in all muscles analyzed, as compared with a single large bolus. This de novo dystrophin expression was able to partially rescue the dystrophic phenotype in treated mdx mice. These data suggest that the repeated use of low doses of naked, unconjugated PMO is likely to be clinically applicable for patients with DMD.
Footnotes
Acknowledgments
This work was supported by the Muscular Dystrophy Campaign, Muscular Dystrophy Ireland, Clinigene (an FP6-EC Network of Excellence), and the UK Department of Health. We gratefully acknowledge the role of the other members of the MDEX Consortium in securing some of this funding, and for helpful input and advice.
Author Disclosure Statement
The authors declare no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.