
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked, lethal genetic disorder affecting the skeletal muscle compartment, and is caused by mutation(s) in the dystrophin gene. Gene delivery of microdystrophin constructs using adeno-associated virus (AAV) and antisense-mediated exon skipping restoring the genetic reading frame are two of the most promising therapeutic strategies for DMD. Both approaches use microdystrophin proteins either directly as a desired construct for gene delivery, using the capacity-limited AAV vectors, or as the therapeutic outcome of gene splicing. Although functionality of the resulting artificial dystrophin proteins can be predicted in silico, experimental evidence usually obtained in transgenic mice is required before human trials. However, the enormous number of potential constructs makes screening assays for dystrophin protein function in vitro and in vivo highly desirable. Here we present data showing that functionality of microdystrophins can be assessed using relatively simple and fast techniques.
Overview Summary
In the present study we wanted to assess fast and efficient methods of evaluating artificial dystrophin proteins with potential therapeutic relevance. We generated four new microdystrophins with large in-frame deletions of the rod domain to be tested for functionality. The four constructs were evaluated in vivo, using intramuscular injection followed by electroporation into tibialis anterior muscle, and in vitro, using a primary-derived immortalized mdx myoblast cell line. All four constructs showed functionality in vivo with correct localization and recruitment of the dystrophin–glycoprotein complex (DGC) proteins to the sarcolemma. All four constructs significantly reduced the number of centronucleated fibers in dystrophin-positive fibers compared with dystrophin-negative (nontransfected) fibers showing functionality at the cellular level with alleviation of the dystrophic pathology. Experiments in which mdx myoblasts expressing the microdystrophin constructs were exposed to hypo-osmotic stress indicated that the constructs protected against membrane damage and recruited the DGC proteins back to the sarcolemma in vitro. On the basis of our results dystrophin functionality can be tested efficiently and consistently by simple methods, which are relatively fast and do not require the need for transgenic mouse models, viral vector production, and viral vector in vivo tests.
Introduction
Dystrophin binds to the dystrophin–glycoprotein complex (DGC), which spans the sarcolemma and connects the extracellular matrix (laminin-α2) with the intracellular cytoskeleton (actin) of muscle fibers through protein–protein interactions mediated by dystrophin; α- and β-dystroglycans; and γ-, α-, β-, and δ-sarcoglycans (Grounds et al., 2005). One of the major mechanical roles of the DGC is to maintain membrane stability and protect the plasma membrane from contraction-induced damage (Ervasti, 2007). Absence of dystrophin results in instability of the sarcolemma and the DGC proteins are reduced in abundance (Turk et al., 2005). The fragile myofibers have increased membrane permeability as evidenced by the uptake of membrane-impermeable dyes such as Evans blue dye (Hamer et al., 2002) and the leakage of muscle-specific proteins such as creatine kinase into the circulation, which can be measured at high levels in serum from patients with DMD (Ozawa et al., 1999). Membrane fragility causes continuous cycles of degeneration and regeneration until exhaustion of the regenerative capacity is reached and the muscle tissue is replaced by fatty infiltrates and fibrosis. Loss of ambulation usually occurs in the early teens and is followed by cardiomyopathy, respiratory failure, and death (Dubowitz, 1987; Engel and Ozawa, 2004).
The only treatment of DMD available today is alleviation of the symptoms with glucocorticoid administration (Angelini, 2007; Chakkalakal et al., 2005). Therefore several treatment strategies aim at correction of the phenotype by replacement of the dystrophin gene (Acsadi et al., 1991; Kochanek et al., 1996), correcting the reading frame of the endogenous gene with antisense oligonucleotides (Lu et al., 2005; Alter et al., 2006), or using myoblast and stem cell transplantation (Torrente et al., 2007; Cerletti et al., 2008).
However, there are major obstacles to the use of gene therapy in DMD. Dystrophin is the largest known mammalian gene with a size of 2.4 Mb, a 14-kb cDNA, and a 427-kDa full-length protein (Hoffman et al., 1987). This gene needs to reach the more than 500 skeletal muscle groups, where the vector and vehicle must efficiently transduce postmitotic tissue and take up long-term residence. In addition, there is a high risk of immunogenicity both toward the vector and toward the therapeutic gene.
Two promising therapies, both being tested in human trials, involve the use of viral vectors for gene delivery to the skeletal muscle compartment, because these vectors have the ability to infect both actively dividing and postmitotic cell nuclei (Xiao et al., 1996; Fisher et al., 1997), and the use of antisense oligonucleotide-mediated exon skipping. Antisense oligonucleotides are targeted against either sequences specifying exon boundaries or exon recognition sequences, thereby excluding one or more exons from the mRNA, reconstituting the reading frame and producing a truncated, but functional protein. This approach has been shown to restore dystrophin expression both in the mdx mouse and in DMD patient myogenic cells (Lu et al., 2005; Aartsma-Rus et al., 2006; van Ommen et al., 2008).
Adeno-associated virus (AAV) has a limited packaging capacity of approximately 5 kb, which is not large enough to accommodate the entire dystrophin cDNA, but AAV has the advantage of long-term persistence within the myonuclei (Fisher et al., 1997). Several studies have provided insight into the functionally essential domains of dystrophin, and those domains that are dispensable in order to make mini- and microdystrophin constructs. The N-terminal actin-binding domain (Corrado et al., 1996; Warner et al., 2002) and the cysteine-rich domain, which binds to the dystroglycan–sarcoglycan complex (Beggs et al., 1991; Rafael et al., 1996), both appear to be required, whereas the C-terminal domain and most of the rod domain can be deleted without severely compromising dystrophin function (Crawford et al., 2000; Harper et al., 2002). Using AAV-mediated expression of microdystrophin, the functionality of several of these constructs has been evaluated in vivo, showing that microdystrophin can protect against contraction-induced injury in mdx muscle (Liu et al., 2005), improve the mdx phenotype (Yoshimura et al., 2004), and alleviate muscular dystrophy in the utrophin/dystrophin double-knockout mouse (Yue et al., 2006).
However, microdystrophin functionality is equally important in relation to antisense-mediated exon skipping as the therapeutic approach. Restoration of the reading frame also occurs spontaneously in some fibers of DMD patients (revertant fibers). Several in-frame-deleted dystrophins have been described in Becker muscular dystrophy patients with a milder clinical phenotype expressing internally deleted, but partly functional dystrophins (Engel and Ozawa, 2004). The dystrophin gene is the largest known mammalian gene with 79 exons, in which (naturally occurring or forced) splicing events could create thousands of different microdystrophin proteins (see the Leiden dystrophin database;
Materials and Methods
Cloning of microdystrophin constructs
Microdystrophin constructs were generated by splicing by overlap extension by the polymerase chain reaction (SOE-PCR), as previously published (Horton et al., 1989), with pBLBecker2 plasmid expressing minidystrophin as a template (Acsadi et al., 1996). For details on the cloning procedure and plasmid map, see Supplementary Table S1a–c and Supplementary Fig. S1 online at
Electroporation of microdystrophin-expressing plasmid into tibialis anterior muscle
Hyaluronidase (0.5 U/μl, 25 μl/muscle) was administered to both tibialis anterior (TA) muscles of 6-week-old mdx mice 2 hr before electroporation. Thirty microliters of one of the four microdystrophin expression constructs (m1–m4) was then injected at a concentration of 1 μg/μl followed by electroporation as previously described (McMahon et al., 2001; Molnar et al., 2004). The mice were killed 10 and 30 days later by cervical dislocation and the TA muscles were harvested and snap-frozen in isopentane cooled in liquid nitrogen for cryosectioning.
Immunohistochemistry
Transverse sections (7 μm) of frozen TA muscles were cut with a cryostat and stained with polyclonal antibodies to dystrophin and β-sarcoglycan (kindly provided by C. Bönnemann, Children's Hospital of Philadelphia, Philadelphia, PA) as previously described (Acsadi et al., 1996; Bönnemann et al., 1996).
Establishment of immortalized mdx myoblast cell line IMMORTO-mdx
Briefly, male H-2Kb-tsA58 mice were obtained from Charles River UK (Margate Kent, UK) and crossed with female mdx mice. Skeletal muscle from the hind limbs of 8-week-old dystrophin-negative male F1 progeny was cut into small pieces and dissociated by stirring in 0.05% trypsin–EDTA in a Wheaton flask. Cells were then plated at low density in supplemented growth medium (Promocell, Heidelberg, Germany) with murine interferon (IFN)-γ (20 U/ml) at 33°C. After 5 days small groups of cells with the myoblast phenotype were picked. From this point on, cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 20% fetal calf serum (FCS) and IFN-γ (20 U/ml) at 33°C. Single-cell clones were then established by limiting dilution. Clones of immortalized myoblasts divided for more than 1 year without losing their ability to fuse into twitching myotubes when differentiated without IFN-γ at 37°C. For each experiment cells from an early passage were used. The protocol used to generate these cells is a modification of that provided by Morgan and colleagues (1994) and has previously been described elsewhere (Brun et al., 2003).
In vivo muscle contractility measurements
Microdystrophin construct m2 was electroporated into tibialis anterior muscle of mdx mice as described previously and muscle contractility was assessed 30 days after treatment (n = 8). The percentage of force deficit was measured after five eccentric contractions at 120 Hz for 30 sec as described by Dudley and colleagues (2004). Phosphate-buffered saline (PBS)-treated mdx mice were used as control (n = 8).
All data are presented as means + 1 SE and a paired Student two-tailed t test was used for statistical evaluation, with p < 0.05 considered significant.
Stable transfection of IMMORTO-mdx cells
IMMORTO-mdx-derived cells (1 × 105) were transfected with 4 μl of GIBCO Lipofectamine Plus (Invitrogen) and 2 μg of each expression vector in 500 ml of Opti-MEM and selected with GIBCO G418 (800 ng/ml; Invitrogen) in DMEM with 20% FBS, glutamine (10 ml/500 ml), and IFN-γ (20 U/ml) (all GIBCO brand; Invitrogen) for 2 to 3 weeks. After clonal expansion the cells were tested for dystrophin expression by Western blotting.
Western blot
Cells were grown to confluence, and allowed to fuse for 8–10 days. Cells were harvested in ice-cold PBS and centrifuged at 1200 rpm for 10 min, and the resulting pellet was lysed in radioimmunoprecipitation assay (RIPA) solution containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40 (NP-40), 0.25% sodium deoxycholate, 1% sodium dodecyl sulfate (SDS), and protease inhibitors (1 nM phenylmethylsulfonyl fluoride [PMSF], 1 mM benzamidine, 5 nM EGTA, leupeptin [0.5 mg/ml], 1 mM Pefabloc, pepstatin [0.7 μg/ml], aprotinin [2 μg/ml]). The lysate was sonicated for 4 sec (Hielscher, Teltow, Germany), centrifuged for 15 min at 4°C at 13 × g, and finally mixed 1:4 with sample buffer (2% SDS, 125 mM Tris-HCl [pH 6.8], 10% glycerol, 5% 2-mercaptoethanol). Forty micrograms of total protein was loaded on 5.5 or 10% bisacrylamide gels (for dystrophin and α-sarcoglycan detection, respectively) and run for 30–45 min at 200 V. Gels were blotted onto a nitrocellulose membrane (Protran BA85; Schleicher & Schuell, Dassel, Germany) for 2–4 hr in Tris–glycine buffer (ICN Biochemicals/MP Biomedicals, Irvine, CA). The membrane was incubated with monoclonal α-sarcoglycan antibody (NCL-a-SARC; Novocastra, Newcastle, UK) diluted 1:100 or with polyclonal dystrophin antibody (Acsadi et al., 1996) diluted 1:200 in Tris-buffered saline (TBS)–Tween with 5% skimmed milk powder overnight at 4°C. The membrane was washed in TBS–Tween, blocked with 5% skimmed milk, and incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit (diluted 1:2000) or HRP-conjugated rabbit anti-mouse (diluted 1:10,000) for 1 hr at room temperature. The reaction was developed with the enhanced chemiluminescence (ECL) detection system (Renaissance enhanced Luminol reagent; PerkinElmer Life and Analytical Sciences, Waltham, MA). The membranes were stripped for 1 hr at 50°C in stripping buffer (100 mM 2-mercaptoethanol, 62.5 mM Tris-HCl [pH 6.7], 2% SDS) followed by reincubation overnight at 4°C with mouse anti-human myosin heavy chain (diluted 1:250). The membrane was washed and then incubated for 1 hr at room temperature with HRP-conjugated rabbit anti-mouse (diluted 1:10,000).
Osmotic shock and creatine kinase measurement
Immortal dystrophin-deficient myoblasts stably expressing microdystrophin constructs, minidystrophin, or no dystrophin (control) were fused for 8–10 days in six-well plates (Falcon; BD Biosciences, San Jose, CA) with 5 × 105 cells per well seeded initially. Fused cells were washed and preincubated with an osmotic solution of 300 mOsm at 27°C (Leijendekker et al., 1996) and then incubated for 15 min with 1 ml of a hypo-osmotic solution of 100 mOsm (10 mM HEPES [pH 7.5], 10 mM KCl, 2 mM MgCl2, 2.4 mM CaCl2, 12 mM NaCl, 1 mM glucose, 50 mM sucrose) or 50 mOsm (5 mM HEPES [pH 7.5], 5 mM KCl, 1 mM MgCl2, 1.2 mM CaCl2, 6 mM NaCl, 0.5 mM glucose, 25 mM sucrose) (Leijendekker et al., 1996). The osmolarity of all solutions before all experiments was confirmed with an osmometer (digital micro-osmometer type 5R; Bachofer, Reutlingen, Germany). Release of creatine kinase (CK) in supernatant was measured with a Granutest 2.5 kit (Merck, Darmstadt, Germany) and normalized to total CK level from fully lysed cells. For each construct, at least two or three individual clones were tested.
Results and Discussion
We designed four new microdystrophin constructs with large in-frame deletions of the spectrin-like repeats of the rod domain according to the structure predictions of dystrophin specified by Cross and colleagues (1990) and Winder and colleagues (1995), and taking into account deletions analyzed previously as described previously. The N-terminal actin-binding domain, hinges 1 and 4 of the rod domain, the β-dystroglycan-binding domain (cysteine-rich domain), and the C-terminal domain were retained in all constructs (Fig. 1A), and thus none of the essential domains were deleted. It has, however, been proposed that the dystrophin rod domain has two functions: binding to the sarcolemmal bilayer and binding of actin through electrostatic interactions (Rybakova et al., 1996; Ervasti et al., 1997) and it was suggested that spectrin-like repeats containing a high number of basic residues mediate the actin binding (Amann et al., 1998). Utrophin, a functional homolog of dystrophin, can completely rescue the dystrophic phenotype of dystrophin-deficient mdx mice (Tinsley et al., 1996) and because utrophin shows no actin-binding activity in the rod domain, this function is probably dispensable in vivo (Tinsley et al., 1998; Amann et al., 1999). More importantly, several DMD patients with in-frame deletions of up to two thirds of the rod domain present with mild clinical Becker muscular dystrophy phenotypes (England et al., 1990; Mirabella et al., 1998), and thus our deletions were not thought to severely compromise dystrophin function.

(
Specifically, our microdystrophin constructs were similar, but not identical, to previously published constructs (Fabb et al., 2002; Harper et al. 2002). These constructs contain slightly different, large in-frame deletions of the rod domain, whereas some constructs carry an additional deletion of most of the C-terminal domain. This type of microdystrophin construct was shown to efficiently prevent or rescue mdx pathology in transgenic mice or after rAAV-mediated delivery (Fabb et al., 2002; Harper et al. 2002; Gregorevic et al., 2006). In contrast, in-frame deletions of the N-terminal actin-binding domain severely reduced the functional capacity of microdystrophin (Banks et al., 2007).
The constructs were cloned into pRcCMV2 and tested for protein expression in transiently transfected 293 cells (data not shown) and stably transfected immortalized mdx myoblasts. The expected molecular weight was then confirmed for proteins extracted from the transfected mdx myoblasts (Fig. 1B). The size of all our constructs allows for packaging into AAV vectors including appropriate regulatory sequences (Dong et al., 1996).
To test the in vivo functionality the four constructs were electroporated as naked plasmid DNA directly into TA muscles of 6-week-old dystrophin-deficient mdx mice. The number of transfected myofibers, using this technique, ranged from 176 ± 34 to 645 ± 224 per TA muscle (Table 1). We then analyzed expression and morphological localization of dystrophin and β-sarcoglycan 10 days postinjection (Fig. 2A–H) and 30 days postinjection (Table 1). All four constructs rescued sarcolemmal dystrophin expression and recruitment back to the sarcolemma of DGC proteins such as β-sarcoglycan, which colocalized with dystrophin (asterisks in Fig. 2A–H). In general, all of the transfected myofibers revealed strong expression of both dystrophin and β-sarcoglycan. In addition, we observed overexpression of (micro-) dystrophin as punctate accumulations in the cytosol of some transfected fibers (Fig. 2I and J). The high number of dystrophin-expressing fibers, which was never observed in control animals of the same age, indicates that the majority of these fibers express transgenic microdystrophin and not naturally occurring revertant dystrophin (Fig. 2K and L; and Lu et al., 2000).

Immunohistochemical detection of dystrophin (green) and β-sarcoglycan (red) in mdx tibialis anterior (TA) muscles transfected with microdystrophin-expressing plasmid. Representative images from transfection with constructs m1 (
Abbreviations: CN, centronucleated; dys, dystrophin; m1–m4, microdystrophin constructs.
Because DGC proteins are strongly reduced in the absence of dystrophin (Turk et al., 2005), thereby rendering the sarcolemma fragile, the recruitment back of these proteins is a strong indicator for dystrophin functionality in vivo. Membrane tears resulting from mechanical stress induced by recurring contractions of the fragile fibers have been proposed by multiple studies to be the key initiator of DMD pathology. This results in disturbed calcium homeostasis and activation of calpains (calcium-activated proteases) capable of cleaving myofibrillar and cytoskeletal proteins and results in focal or complete lysis of the damaged fibers (Gissel, 2005). This has led to suggestions that any therapy stabilizing the muscle cell membrane could rescue the dystrophic pathology (Tinsley et al., 1998; Squire et al., 2002; Bentzinger et al., 2005). However, data suggest that dystrophin may also act directly in cell signaling (Chakkalakal et al., 2006) and it has been shown that dystrophin interacts either directly or indirectly with calcium channel proteins (Friedrich et al., 2008), suggesting that full dystrophin function cannot be easily replaced by any stabilizing protein (Batchelor and Winder, 2006). Therefore it is essential to restore dystrophin expression even if only with a partially functional protein. It has been reported that Becker muscular dystrophy patients, who express partially functional dystrophin, present with a milder phenotype and these patients have a much improved quality of life and life expectancy compared with Duchenne muscular dystrophy patients (Engel and Ozawa, 2004).
Expression of the microdystrophin constructs also resulted in a statistically significant reduction in the number of centronucleated myofibers when comparing dystrophin-positive and -negative fibers after electroporation of naked plasmid DNA constructs into TA muscle (Table 1, using Student two-tailed t test; p < 0.05 for all constructs 10 and 30 days after treatment except for m2, for which p = 0.08 thirty days after treatment), indicating alleviation of the dystrophic pathology. Adult mdx mice generally present with a high number of centrally nucleated (CN) myofibers, about 50–60% in young adults, corresponding to the numbers from dystrophin-negative fibers presented in Table 1, up to as high as >90% in aged mdx mice (Gilbert et al., 2003; Marquez et al., 2007), whereas the CN index remains at about 1% in wild-type control mice (Marquez et al., 2007). These centrally nucleated fibers are considered a result of active regeneration and represent either immature, newly forming myotubes before maturation or regenerated fibers not capable of full maturation. The latter is more likely because these mice display active regeneration from 3 to 8 weeks of age (Turk et al., 2005). The reduction in CN number in adult muscles expressing the microdystrophins suggests either a relocation of the nuclei to the periphery mediated by signaling from dystrophin in transfected myofibers, or these could be newly regenerated and matured fibers originating from satellite cells expressing dystrophin de novo. In addition, we also performed muscle contractility measurements 30 days after treatment with microdystrophin constructs, and we observed a significant difference in percentage force drop between the assessed construct (m2) and saline-injected control mice (Fig. 3). These results, showing an improvement in muscle physiology, corroborate the amelioration of the mdx phenotype as observed by CN reduction.

Muscle contractility measurements of mdx tibialis anterior muscles 30 days after treatment with microdystrophin construct m2 (solid column) or PBS control (open column) (n = 8 for each group) showed a significant improvement in resistance (* p < 0.05). Data are presented as mean (column) + 1 SE (error bar).
To further analyze our microdystrophin constructs we established primary derived immortalized mdx myoblasts by isolating myoblasts from a cross between mdx mice and the H-2Kb-tsA58 transgenic mice as previously described (Morgan et al., 1994; Brun et al., 2003). These IMMORTO-mdx cells were then stably transfected with the four constructs and minidystrophin (Becker-type dystrophin). The stably transfected cells were induced to differentiate and fuse and the expression of α-sarcoglycan, and myosin heavy chain was measured by Western blotting (Fig. 4). There was increased expression of α-sarcoglycan in the cells stably expressing any of the four constructs or minidystrophin compared with nontransfected control cells (mdx). These results thus confirm our in vivo findings with the recruitment of DGC proteins to the sarcolemma.

Primary derived IMMORTO-mdx myoblasts were transfected with Becker dystrophin (mini) (B-Dys), the microdystrophin constructs (m1–m4), vector alone (pRcCMV2), or no dystrophin (mdx). The cells were then induced to fuse and expression of α-sarcoglycan (50 kDa) was detected by Western blot. Myosin heavy chain (MHC, 200 kDa) was detected on the same membrane to confirm differentiation and as a loading control for amount of protein. Cells from the transfection with m3 microdystrophin before fusion (unf) were included to verify specificity of the staining because α-sarcoglycan and myosin heavy chain are not present in proliferating myoblasts.
The relative membrane stability of mdx myoblasts expressing the four constructs, minidystrophin, or no dystrophin (mdx) was assessed directly, using osmotic shock by treating the cells in hypo-osmotic solutions followed by measurement of creatine kinase release into the medium (Fig. 5). All four constructs protected against shock-induced membrane damage and decreased CK release in a manner similar to minidystrophin compared with nontransfected mdx myoblasts (p < 0.01). In addition, all the constructs protected the membranes almost as effectively as normal, full-length dystrophin in wild-type cells. CK release is used as a measure in clinical assessment of muscular dystrophy patients and the decrease in CK release from the cytosol of mdx myoblasts expressing the microdystrophins indicates that microdystrophin is fully capable of rescuing membrane stability, thereby protecting the myofibers against stress.
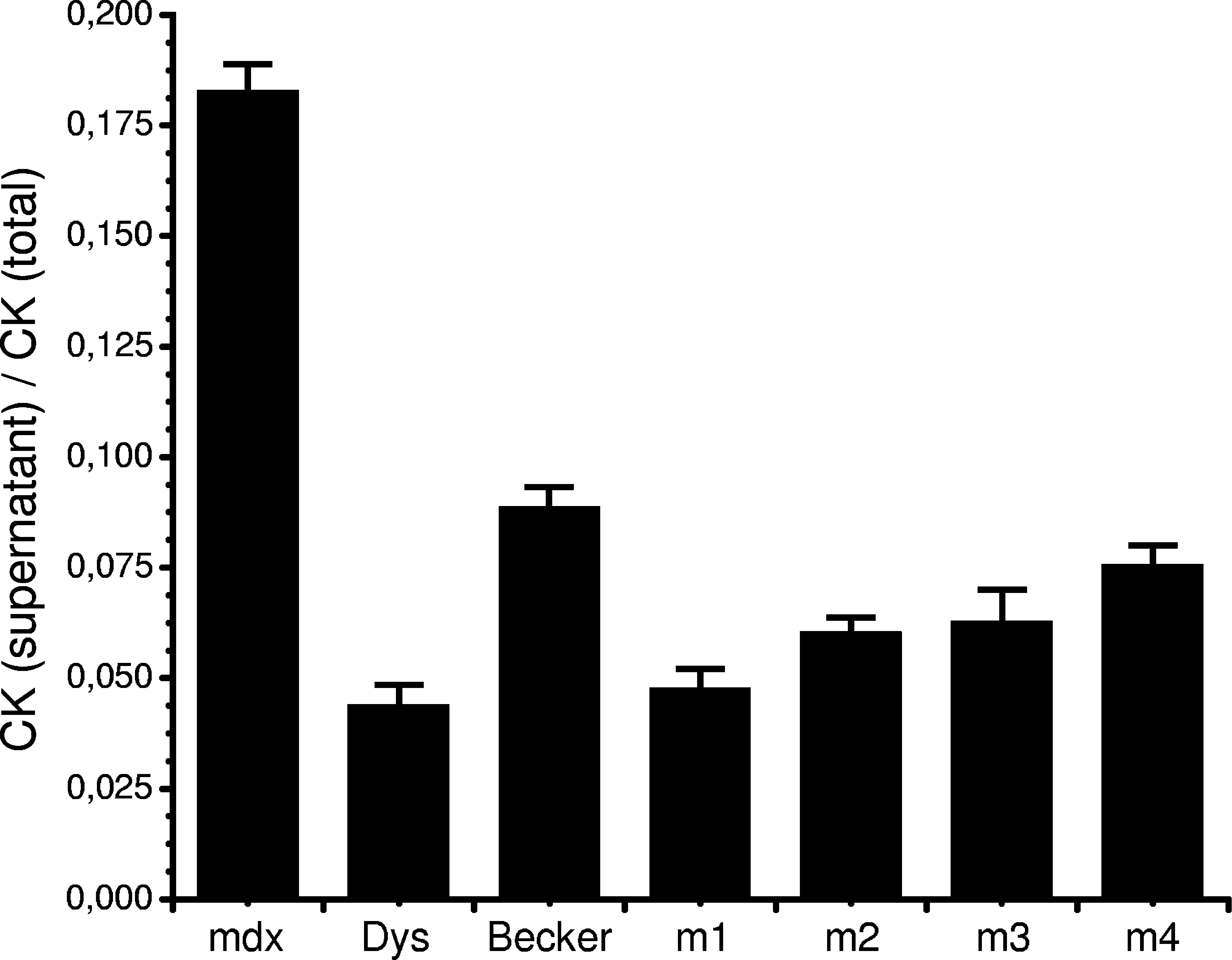
Primary derived IMMORTO-mdx myoblasts were transfected with Becker dystrophin (mini) (Becker, n = 35), the microdystrophin constructs (m1, n = 51; m2, n = 25; m3, n = 18; m4, n = 44), or no dystrophin (mdx, n = 45). Wild-type IMMORTO myoblasts expressing full-length dystrophin were included as positive control (Dys, n = 38). The cells were allowed to fuse for 8–10 days before the myotubes were subjected to stress by incubation in a hypo-osmotic solution (100 mOsm) and creatine kinase (CK) release into the medium was measured. CK release in the medium (supernatant) was divided by total CK release from lysated myotubes (total). For each construct, wild-type and mdx control cells of at least three individual clones were tested. n = total number of measurements for each construct and control. Data are presented as mean (column) + 1 SE (error bar).
In this study we used various assessment methods to test our microdystrophin constructs, and we showed both that our constructs were functional and that this could be experimentally evaluated using simple electroporation of plasmid DNA in vivo and osmotic shock in vitro.
This easy, fast, and cost-efficient functional assessment may be valuable for several reasons. First, many studies that have analyzed microdystrophins or other candidates for gene therapy of muscular dystrophy have used transgenic mouse models. In these models the transgene of interest is expressed early during embryonic development or fetal life, which may not accurately model a therapeutic scenario in which the therapeutic gene is likely to be introduced later. The effect of the therapeutic gene may be diminished once the pathology has progressed further and the immune system is more mature after birth. Therefore, the risk of an immune response, both toward the transgene itself and the vector, will be higher if introduced into a patient with a mature immune system. It has been reported that dystrophin-positive and -negative canine skeletal muscles elicit a massive immune response against the AAV vector (Wang et al., 2007; Yuasa et al., 2007). In addition, myoblasts, myotubes, and immature, regenerating fibers can act as antigen-presenting cells themselves, thus providing a means for the immune response to react against the transgene (Wiendl et al., 2005). This ability has been suggested to partly account for an enhanced immune response toward transgene expression in mdx mice (Yuasa et al., 2002).
Our functional screen also removes the need for production of viral vectors for construct functionality testing. Viral production time can be up to 4 months and there is a constant need for amplification, purification, and titer assessment for efficient transduction. On the other hand, the in vivo setup presented here is easily done by conventional bacterial cloning followed by injection, electroporation, and assessment.
In conclusion, these are screening assays and further testing for safety and efficacy of the therapeutic approach remains necessary (e.g., animal models and human trials). However, these assays significantly reduce the vast number of microdystrophin constructs to be screened for potential patient use.
Footnotes
Acknowledgments
The authors thank Carol Allen, Stephen Prescott (Montreal), and Ursula Klutzny (Munich) for technical assistance. The authors also thank George Karpati and Josephine Nalbantoglu for helpful comments on the manuscript. R.S., C.T., M.C.W., and H.L. are members of the German Network on Muscular Dystrophies (MD-NET, 01GM0601), funded by the German Ministry of Education and Research (BMBF, Bonn, Germany). MD-NET is a partner of TREAT-NMD (EC, 6th FP, proposal 036825). This project was further funded by grants from the Deutsche Forschungsgemeinschaft (DFG) and the German Duchenne Parents Project (Action Benni and Co.) to H.L. and by a grant from the Lundbeck Foundation (R19-A2177) to L.H.J.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.