
Abstract
Numerous strategies are under development for the correction of deleterious effects of mutations in muscular dystrophies, and these strategies must be validated in compelling models. Cellular models seem straightforward to set up; however, the proliferative capacity of muscle cells isolated from dystrophic patients is limited, and in addition it is difficult to envisage the use of large muscle biopsies from patients to obtain enough cells for ex vivo assessments. To overcome these problems, we have devised a strategy to obtain, from a patient with Duchenne muscular dystrophy (DMD), an inexhaustible source of myogenic progenitor cells with a deletion of exons 49 and 50 in the dystrophin gene. Starting material consisted of dermal fibroblasts isolated from a skin biopsy taken in a noninvasive way. These fibroblasts were first immortalized by telomerase gene transfer. Subsequent cell lines were converted into myogenic cells by means of a lentiviral vector encoding an inducible MyoD construct. Before myogenic induction, engineered DMD fibroblasts were able to proliferate infinitely. Under induction conditions, they were converted into myogenic cells, which differentiated into large multinucleated myotubes. We used these DMD fibroblast cell lines to assess dystrophin rescue by using engineered U7 small nuclear RNAs harboring antisense sequences required to restore an in-frame dystrophin mRNA by skipping exon 51. Further molecular analyses showed dystrophin rescue ex vivo as well as in vivo after engrafting of treated cells into regenerating muscles in immunodeficient mice.
Introduction
The development of new therapies for muscular dystrophies has reached a new momentum and a number of new strategies have been developed to correct the deleterious effects of the mutations. While gene therapy has focused primarily on gene replacement, alternative approaches targeting RNA have emerged, either to downregulate gene transcription or to modulate the processing of target pre-mRNA transcripts, such as in the exon-skipping approach (Goyenvalle et al., 2004; van Deutekom et al., 2007). Pharmacological approaches using specific drugs also provide potential therapeutic avenues as shown by a report using the drug PTC124 (Ataluren), which promotes readthrough of nonsense mutations (Welch et al., 2007).
All these therapeutic strategies need to be validated in animal or cellular models. Mice with spontaneous mutations that mimic the human muscle disorders are rare and the sequences of interest are often different from those found in the human disease. Transgenic mice expressing the human mutations can also be made and used to validate strategies, although the mutation and/or the mutated mRNA remain in a murine context. Several transgenic mice have now been developed to investigate the physiopathology of different human muscular dystrophies. However, such models do not exist yet for every mutation, particularly for diseases such as Duchenne muscular dystrophy (DMD), for which numerous mutations have been described (Deburgrave et al., 2007).
For such diseases, models should be tailored for each mutation in order to assess the efficiency of strategies such as exon skipping or stop codon readthrough. Human primary muscle cell cultures isolated from the muscle of dystrophic patients represent a good model because the mutation is found in its natural genomic environment and is expressed in a human cellular context. Cellular models are easy to set up and manipulate for screening therapeutic molecules. However, two major difficulties hamper the use of primary human muscle cell cultures: the accessibility and availability of muscle biopsies from patients affected with muscular dystrophies, and the limited proliferative capacity of adult human myoblasts. Human myoblasts, like all somatic cells, enter into replicative senescence after a finite number of divisions that is inversely correlated with the age of the donor (Hayflick, 1965; Renault et al., 2000). In addition, excessive proliferation of the satellite cells in muscular diseases involving continuous cycles of degeneration and regeneration such as DMD, leads to the rapid exhaustion of their regenerative capacity: the in vitro proliferative capacity of myoblasts isolated from patients with DMD was found to be considerably less than that of aged-matched controls (Webster et al., 1986; Renault et al., 2000).
In this study, we have developed an innovative cellular model for the validation of gene therapy or pharmacological approaches for muscular dystrophies. Fibroblasts were isolated from a skin biopsy of a patient with DMD, immortalized to generate a cell line, and then transduced with an inducible MyoD construct. Under nonpermissive conditions these immortalized fibroblasts are able to proliferate infinitely, and can thus be amplified. Under permissive conditions, these modified cells are converted to a muscle fate and express a myogenic program. Because the mutation affecting the patient with DMD was an out-of-frame deletion in the dystrophin gene, an exon-skipping strategy was designed to restore the reading frame. Using a lentiviral vector expressing antisense sequences linked to a modified U7 small nuclear RNA (snRNA), we show that the converted cells produce new in-frame truncated dystrophin transcripts. Moreover, these cells were transplanted into regenerating muscles of immunodeficient mice and, although they were less efficient in participating to the regeneration of the host's muscle than human myoblasts, human dystrophin proteins were detected at the periphery of the muscle fibers containing human nuclei originating from the injected fibroblasts.
Materials and Methods
Cell cultures
Primary fibroblast and myoblast cell cultures were established from explants of skin and biceps, respectively, as described previously (Mouly et al., 1993), in accordance with French and Italian ethics legislation. Fibroblasts were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) whereas myoblasts were grown in Ham-F10 supplemented with 20% FBS. The mean population doubling (MPD) at every passage was calculated according to the formula log N/log 2, where N is the number of cells at the time of passage divided by the number of cells initially attached after seeding. To induce differentiation, both types of cultures were switched to DMEM with transferrin (100 μg/ml) and insulin (10 μg/ml; Sigma-Aldrich, St. Louis, MO). For myoconversion, doxycyclin (2 μg/ml; Sigma-Aldrich) was added in the differentiation medium. All cultures were grown in humidified incubators at 37°C in 5% CO2.
Cell transduction
For immortalization, the fibroblasts were transduced with a pBABE-puro-based retroviral vector containing sequence encoding the catalytic subunit of human telomerase (hTERT) as previously described (Auré et al., 2007) and then selected in the presence of puromycin (1 μg/ml) for 10 days. Transduction experiments using lentiviral vectors were performed overnight in the presence of Polybrene (4 μg/ml; Sigma-Aldrich). The three lentiviral vectors used in this study contained (1) enhanced green fluorescent protein (eGFP) under the control of the cytomegalovirus (CMV) promoter, (2) murine MyoD under the control of the Tet-on inducible construct (Barde et al., 2006), or (3) a modified U7 snRNA construct promoting the antisense sequence CCUCUGUGAUUUUAUAACUUGAUUCAAGGAAGAUGGCAUUUCU directed against exon 51 of the human dystrophin mRNA (Benchaouir et al., 2007). Transduction efficiency of the immortalized fibroblasts was tested with a lentiviral vector encoding eGFP and more than 96% of the cells expressed GFP, using a multiplicity of infection (MOI) of 5 (i.e., using 500,000 transducing units [TU] of vectors to transduce 100,000 cells) without vector-associated cytotoxicity (data not shown). The lentiviral vector titer (TU/ml) was determined by quantitative real-time polymerase chain reaction (PCR) as described elsewhere (Charrier et al., 2005).
RT-PCR
Total RNA was extracted from cell cultures with TRIzol reagent (Invitrogen). One microgram of RNA was reverse transcribed into cDNA, using murine leukemia virus (MLV) reverse transcriptase (RT), and RT products were subjected to PCR amplification with 2 × ReddyMix reagent (Abgene, Epsom, Surrey, UK). The following amplification conditions using specific primers were applied: 1 min at 95°C, 1 min at 60°C (MyoD) or 55°C (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]), and 1 min at 72°C (35 cycles for MyoD and 25 for GAPDH). Amplification products were separated on a 1.5% agarose gel containing ethidium bromide for visualization, and quantification was performed with Gel Doc 2000 software (Bio-Rad, Hercules, CA). For dystrophin detection, 300 ng of total RNA was used for RT-PCR analysis, using the Access RT-PCR system (Promega, Madison, WI) in a 50-μl reaction using external primers 46F-AGGAAGCAGATACATTGCT and 53R-TTTCATTGAACTGTTGCCTC. cDNA synthesis was carried out at 45°C for 45 min, directly followed by the primary PCR of 30 cycles of 94°C (30 sec), 58°C (1 min), and 72°C (2 min). Two microliters of each of these reactions was then reamplified in nested PCRs by 28 cycles of 94°C (30 sec), 58°C (1 min), and 72°C (2 min), using the internal primers 47F-(TTACTGGTGGAAGAGTTGCC) and 52R-(TGATTGTTCTAGCCTCTTGA). PCR products were analyzed on 2% agarose gels. The 439-bp product was purified and analyzed by DNA sequencing.
Immunochemistry
Cell cultures were fixed in ethanol, blocked in 1% FBS, and then incubated with monoclonal antibodies against MyoD (MyoD1, diluted 1:50; Dako, Carpinteria, CA) or myosin heavy chains (MF20, diluted 1:20; Developmental Studies Hybridoma Bank [DSHB], University of Iowa, Ames City, IA). Cryosections of tibialis anterior (TA) muscle were analyzed with antibodies against human lamin A/C (NCL-LAM-A/C, diluted 1:400; Novocastra Laboratories, Newcastle upon Tyne, UK), human spectrin (NCL-SPEC1, diluted 1:50; Novocastra Laboratories), and human dystrophin (NCL-Dys3, diluted 1:5; Novocastra Laboratories). Specific antibody binding was revealed with Alexa Fluor 488 directly coupled to the secondary antibody (Invitrogen Molecular Probes, Eugene, OR). To visualize nuclei, slides were mounted in Mowiol containing Hoechst (10 μg/ml; Sigma-Aldrich). All pictures were taken and digitalized with an Olympus BX60 microscope and MetaVue software (Molecular Devices, Sunnyvale, CA).
Cell transplantation
Human cells (1 × 106) were injected in the cryodamaged TA muscles of Rag2−/−γC−/− immunodeficient mice as described previously (Cooper et al., 2003). Two TA muscles were injected with human control myoblasts, five TA muscles with U7ex51-Ind.MyoD-hTERT-DMD fibroblasts, five TA muscles with Ind.MyoD-hTERT-DMD fibroblasts, and two TA muscles with hTERT-DMD fibroblasts. To induce the Tet-on promoter, mice were intraperitoneally injected with doxycyclin (40 μg/g) daily from day 4 to day 9 after cell injection, and then fed with doxycyclin water (200 μg/ml) containing 2.5% sucrose. One month after the injection of cells, mice were killed and injected TA muscles were mounted in 6% gum tragacanth (Sigma-Aldrich), frozen in liquid nitrogen-cooled isopentane, and stored at −80°C.
Results and Discussion
Generating an immortalized DMD fibroblast cell line from skin
Skin and muscle biopsies were performed on a 14-year-old DMD patient with a deletion in exons 49 and 50, and primary cultures of both skin fibroblasts and muscle cells were established. As shown in Fig. 1A, the myoblast culture stopped dividing after nine mean population doublings (MPDs), when the DMD myoblasts entered into proliferative senescence. A reduced proliferative capacity of DMD myoblasts was also measured in two other primary cultures isolated from muscle biopsies from 7- and 11-year-old DMD patients, and proliferative arrest was reached at 16 and 12 MPDs, respectively, whereas an aged-matched control culture made 30 MPDs, in agreement with previous studies (Renault et al., 2000). The DMD fibroblasts stopped proliferating after 29 MPDs (Fig. 1B), confirming that the proliferative capacity of the DMD fibroblasts is less altered than that of myoblasts.

(
It was demonstrated previously that expression of telomerase (hTERT) in human fibroblasts leads to their immortalization, in contrast to human myoblasts (Bodnar et al., 1998; Di Donna et al., 2003). DMD fibroblasts transduced with a retroviral vector containing the cDNA encoding the catalytic subunit of human telomerase cDNA (hTERT) were able to make more than 100 MPDs and were considered to be immortalized (Fig. 1B). Therefore, we have demonstrated that a primary culture of human DMD skin fibroblasts can be easily established from a noninvasive superficial skin biopsy and immortalized, thus giving rise to an unlimited source of cellular material.
Inducible forced MyoD expression in immortalized fibroblasts leads to converted muscle cells
Ectopic expression of MyoD in some nonmuscle cells is able to activate the myogenic program and force myogenesis in some nonmyogenic cells including fibroblasts, which then withdraw from the cell cycle, differentiate, and fuse to form multinucleated myotubes (Lattanzi et al., 1998). As shown by Cooper and colleagues, these converted fibroblasts express a wide range of muscle-specific genes associated with muscular dystrophies (Cooper et al., 2007). In our study, a lentiviral vector encoding an “all-in-one” Tet-on inducible MyoD construct was used to transduce the immortalized DMD fibroblasts. Under nonpermissive conditions, the Ind.MyoD-hTERT-DMD fibroblasts were still able to proliferate. When doxycyclin was added to the culture medium, an induction of MyoD mRNA was already detected after 2 hr by RT-PCR (Fig. 2A). Expression of MyoD was observed by immunofluorescence 24 hr after doxycyclin induction in 85 ± 7% of the cells, thus confirming the high transduction efficiency and reliable induction of MyoD under permissive conditions (Fig. 2B). After induction of MyoD under proliferating conditions, the number of dividing cells, identified by Ki-67 detection, decreased from 66% at 24 hr postinduction, to 10% 4 days postinduction (data not shown).

Forced MyoD expression in immortalized DMD fibroblasts leads to converted muscle cells. Doxycylin was added to Ind.MyoD-hTERT-DMD fibroblasts cell cultures and MyoD was rapidly induced as shown by (
To determine whether MyoD could induce myogenesis in immortalized DMD fibroblasts and induce myotube formation, cultures were allowed to reach confluence and then switched to differentiation medium supplemented with doxycyclin. As shown in Fig. 2C, the cells were able to fuse and form multinucleated myotubes. Expression of muscle-specific proteins such as myosin heavy chain was confirmed by immunofluorescence analysis using the MF20 antibody. The fusion index was 67 ± 7% after 10 days in differentiation medium, a value close to the 87 ± 5% measured in a primary culture of human myoblasts used as a control. It should be noted that, once induced, MyoD is still expressed after 6 days of differentiation (data not shown). As shown previously for fibroblasts with noninducible forced MyoD expression, the Ind.MyoD-hTERT-DMD fibroblasts described here are able to differentiate and form myotubes under permissive conditions. However, in contrast to MyoD-transfected fibroblasts that rapidly withdraw from the cell cycle, the Ind.MyoD-hTERT-DMD fibroblasts can still be expanded under nonpermissive conditions and differentiated when needed by inducing MyoD and differentiation.
Testing an exon-skipping approach to restore dystrophin in DMD converted muscle cells
The mutation affecting the DMD patient corresponds to a deletion of exons 49 and 50 in the dystrophin gene. This deletion creates a disruption of the open reading frame (ORF), resulting in the absence of the dystrophin protein that is associated with a severe DMD phenotype. However, the dystrophin reading frame can be restored if exon 51 is skipped. The absence of exons 49 to 51 produces a novel in-frame dystrophin mRNA that can be translated into a truncated quasi-dystrophin protein. Thus, the del-49-50 mutation is potentially eligible for exon skipping. The Ind.MyoD-hTERT-DMD fibroblast cell line carrying this mutation has been used to test antisense sequences promoting the skipping of exon 51.
A lentiviral vector expressing antisense sequences aiming at skipping exon 51 linked to a modified U7 small nuclear RNA was produced as described previously (Benchaouir et al., 2007). Ind.MyoD-hTERT-DMD fibroblasts were transduced with this U7ex51 vector, amplified, and converted to myogenesis to evaluate the efficiency of the antisense approach. After 10 days of differentiation, expression of dystrophin was measured by RT-PCR and compared with that of nonskipped cells. As seen in Fig. 3, a 672-bp product corresponding to the mutated dystrophin was amplified from the nonskipped converted DMD cells whereas a 439-bp product amplified from the skipped mRNA was detected in converted DMD cells expressing the modified U7ex51 construct. Note that an additional band between the nonskipped and skipped products is visible. This is due to heteroduplex formation and has been described previously (Aartsma-Rus et al., 2003). DNA sequencing of the 439-bp product, which appears to be the major species detected in treated cells, confirmed the absence of exons 49, 50, and 51 in the novel dystrophin transcripts (data not shown). This result demonstrates that exon 51 antisense sequence delivered by the modified U7ex51 construct is able to efficiently provoke skipping of exon 51 of the dystrophin mRNA, generating a new in-frame exon 48–52 dystrophin mRNA in converted Ind.MyoD-hTERT-DMD fibroblasts.
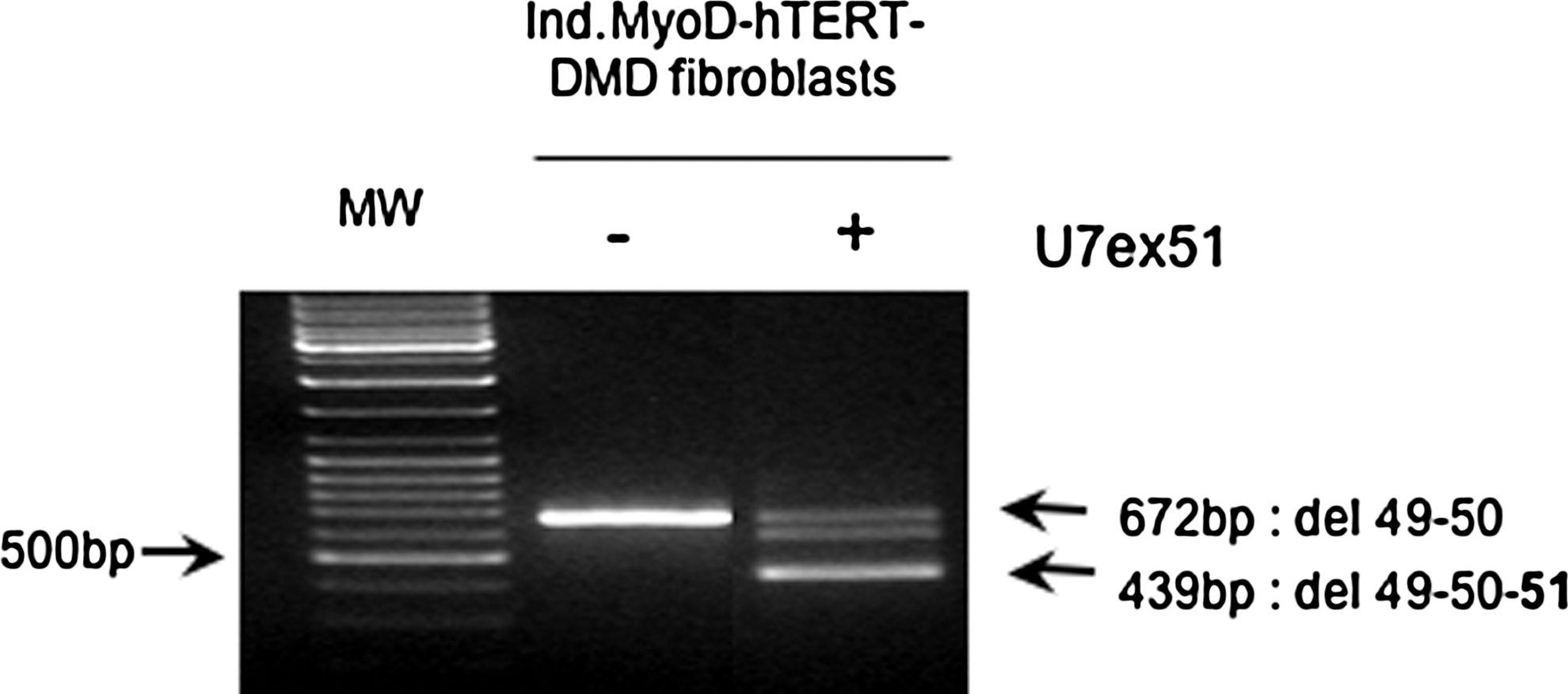
Skipping of exon 51 restores new in-frame dystrophin transcripts in DMD converted muscle cells. The Ind.MyoD-hTERT-DMD fibroblast cell culture was transduced with a lentiviral vector that delivers an exon 51 antisense sequence linked to a modified U7 snRNA (U7ex51) and then converted in muscle cells for 10 days. Expression of the dystrophin transcripts was analyzed by RT-PCR.
In vivo recovery of human dystrophin in DMD muscle fibers
To determine whether the generation of skipped transcripts leads to the restoration of functional dystrophin protein, U7ex51-Ind.MyoD-hTERT-DMD fibroblasts amplified without doxycyclin were transplanted into cryodamaged tibialis anterior (TA) muscle of immunodeficient Rag2−/−γC−/− mice to participate in muscle regeneration (Cooper et al., 2003), along with Ind.MyoD-hTERT-DMD fibroblasts and hTERT-DMD fibroblasts as controls. Each type of cell was injected into one leg, whereas human control (i.e., non-DMD) myoblasts were used as a positive control in the contralateral leg. To visualize human fibers within the murine host muscle, immunofluorescence staining was performed on sections, using specific monoclonal antibodies directed against human spectrin to detect human fibers and nuclear human lamin to detect human nuclei (Fig. 4). Serial sections were stained with a monoclonal antibody specific for human dystrophin. As revealed by human spectrin immunostaining in muscles transplanted with Ind.MyoD-hTERT-DMD fibroblasts (Fig. 4E) compared with those injected with hTERT-DMD fibroblasts (Fig. 4G), the Ind.MyoD construct could convert the fibroblasts to myogenesis in vivo. Although this study is not quantitative, we observed that the number of fibers expressing human proteins and/or containing human nuclei formed by Ind.MyoD fibroblasts was always lower than that observed with human myoblasts (e.g., 35–90 fibers with Ind.MyoD fibroblasts vs. up to 300 fibers with myoblasts). No human dystrophin immunostaining was detected in muscles transplanted with converted DMD muscle cells (Fig. 4F), as expected, because the deletion leads to the absence of dystrophin in human fibers. In contrast, human dystrophin was detected in the muscle transplanted with human control myoblasts (Fig. 4B) but also with converted U7ex51-DMD muscle cells (Fig. 4D). As can be seen on Fig. 4A and C in panel I, the same positive dystrophin fibers were also stained with human spectrin on the serial section, confirming the human status of these fibers. Because the restored dystrophin is detected at the periphery of fibers as typically observed for full-length dystrophin, it suggests that the novel skipped dystrophin is able to reach its location under the membrane and participate to the dystrophin–glycoprotein complex. This in vivo recovery of correctly located dystrophin in DMD muscle fibers confirms the efficiency of the exon-skipping approach, and validates our new cellular model to assess gene therapy approaches.

The skipped dystrophin proteins in U7ex51-DMD muscle fibers are located under the membrane. Ind.MyoD-hTERT-DMD fibroblasts transduced with U7ex51 (
In conclusion, this study proposes an innovative approach to generate new cellular tools to evaluate therapeutic approaches for muscular dystrophies. Because skin biopsies are less invasive for patients than muscle biopsies, we have isolated skin fibroblasts from a patient with DMD to engineer an immortalized cell line that, under permissive conditions, can be converted to muscle cells and express muscle-specific proteins. Such cell lines could be easily generated for any neuromuscular disease and provide the opportunity to easily assess pharmacological or gene therapy approaches on human cells containing the mutation in its natural context. In this short report, an exon-skipping approach was successfully tested to overcome the deletion of dystrophin exons 49 and 50 as a proof of principle for using Ind.MyoD-hTERT fibroblast cell lines to evaluate therapeutic strategies. In addition, because this cell line has no proliferation limits, amplification of these modified fibroblasts can be performed in order to obtain a large number of cells, which can then be converted for high-throughput screening.
Footnotes
Acknowledgments
The authors acknowledge the patients with DMD for biopsies, W.E. Wright and Geron Co for sharing hTERT constructs, J. Riederer and L. Dollé for technical support, and the following financial sources of support: Association Française contre les Myopathies (AFM), Université-Paris 6, INSERM, CNRS, and MYOAMP Strep (EC, 6th FP, LSHB-CT-2006-037479).
Author Disclosure Statement
No competing financial interests exist.