
Abstract
Effective techniques for the stable genetic modification of peripheral T cells would facilitate functional gene studies and the development of gene therapeutic approaches. However, many approaches to genetically modify T cells are hampered by low transfection efficiency, direct cell toxicity, and the need for specialized laboratory space. In this study we investigated the Amaxa Nucleofector platform, a nonviral technique to transfect primary human T cells. A plasmid equipped with two different promoters enabled concomitant expression of a gene of interest and of a cell surface marker allowing for immunomagnetic cell enrichment. This resulted in highly purified populations of gene-modified T cells and, after repeated enrichment steps, provided stably and homogeneously transfected, fully functional human T cells. In summary, this study provides proof of principle that human T cells can be altered to homogeneously and stably express a gene of interest by a nonviral technique. This should enable further studies on T cell physiology and ultimately facilitate the translation of treatment approaches either for diseases that are caused by defective gene function in T cells or for diseases that require genetically designed T cell therapy.
Introduction
Amaxa Nucleofector technology (Lonza, Cologne, Germany) is a modified electroporation method for delivering DNA directly into the nucleus even of difficult-to-transfect cells (Hamm et al., 2002; Gresch et al., 2004; Schakowski et al., 2004). It has been shown that nucleofection is a suitable method by which to transfect cells of hematopoietic origin (Lai et al., 2003; Goffinet and Keppler, 2006). More recently it has been described that nucleofected and enriched human T cells differentiate into helper T type 1 (Th1) and helper T type 2 (Th2) cells similar to nontransfected control cells, but could not demonstrate stable expression of the desired transgene (Tahvanainen et al., 2006).
To study gene function it is necessary to achieve long-term expression of the transgene in a homogeneous cell population. As transfection efficiency is typically far below 100% and chromosomal integrations of the transgene are rare events using nonviral techniques, efficient purification of stable transfectants is indispensable. Commonly, vectors expressing fluorescent marker proteins such as green fluorescent protein (GFP) are used to select for positive transfectants by fluorescence-activated cell sorting (FACS). This method is, however, time-consuming and associated with high financial costs. Enrichment with immunomagnetic beads after ectopic coexpression of a surface molecule otherwise not expressed on the cells represents an alternative approach allowing for easy and high-throughput isolation of transfected cells (Schneider and Rusconi, 1996; Gaines and Wojchowski, 1999). For this purpose C-terminal truncated versions of the low-affinity nerve growth factor receptor (ΔLNGFR) and mouse MHC class I molecule (H-2Kk) have been established both for detection and purification of transfected cells (Holtkamp et al., 1981; Fehse et al., 1997). Coupling expression of the gene of interest with a selection marker can be achieved by cotransfection of two different plasmids, originating from a single plasmid by bicistronic expression, using an internal ribosomal entry site or by dual expression driven by two different promoters (Yu et al., 2003).
Here we describe an easy and reliable strategy for the generation and purification of stably transfected primary human T cells exhibiting genomic integration and homogeneous long-term expression of a gene of interest. We demonstrate that nucleofection is a suitable method for transfection of resting primary human T cells. A dual-expression plasmid allows for coexpression of a gene of interest and a selection marker for immunomagnetic purification. This provides an efficient method not only for studying gene functions in primary human T cells but also potentially for therapeutic application of genetically modified T cells.
Materials and Methods
Cell isolation
Peripheral blood mononuclear cells (PBMCs) obtained from healthy donors were isolated from buffy coat by density gradient centrifugation over Ficoll-Hypaque 1077 (Pharmacia, Uppsala, Sweden). PBMCs were washed twice in phosphate-buffered saline (PBS) and subsequently used for magnetic cell sorting. CD4+ cells were purified by depletion of non-CD4+ T cells with the CD4+ T cell isolation kit II from Miltenyi Biotec (Bergisch Gladbach, Germany). Purity was routinely between 95 and 98% as determined by flow cytometry.
Plasmids
The eukaryotic dual-expression vector pCDH-CMV-MCS-EF1-copGFP containing two independent internal promoters (i.e., cytomegalovirus [CMV] and elongation factor-1α [EF1] promoters) was purchased from System Biosciences (Mountain View, CA). The coding sequence of C-terminal truncated low-affinity nerve growth factor receptor (ΔLNGFR) was cloned from the pMACS-LNGFR plasmid (Miltenyi Biotec) by polymerase chain reaction (PCR) amplification, using the following primers: forward primer, 5′-GCGGCTAGCGCCACCATGGGGGCAGGTGCCACCG-3′; and reverse primer, 5′-ATAGCGGCCGCTCACCTCTTGAAGGCTATGTAG-3′. The PCR fragment encoding ΔLNGFR was digested with NheI/NotI and ligated into the NheI/NotI-digested plasmid pCDH-CMV-MCS-EF1-copGFP, yielding the plasmid pCDH-CMV-LNGFR-EF1-copGFP. The plasmid pCDH-CMV-GFP-EF1-LNGFR was cloned according to the following strategy: The DNA fragment EF1–copGFP from the original plasmid was removed with NotI and SalI. The EF1α promoter was reintroduced into the NotI/SalI sites with an additional PmeI site downstream of the EF1α promoter, using the following primer pair: forward primer, 5′-GATCCGCGGCCGCAAGGATCTG-3′; and reverse primer, 5′-GCGGTCGACGTTTAAACGTCTAGCGTAGGCGCCGGTCACAG-3′, creating pCDH-CMV-MCS1-EF1-MSC2. copGFP (copepod Pontellina plumata GFP) was amplified with forward primer 5′-GCGTCTAGAACGCCACCATGGAGAGCGAC-3′ and reverse primer 5′-ATAGCGGCCGCTTAGCGAGATCCGGTGGAG-3′ and ligated into the XbaI/NotI sites of pCDH-CMV-MCS1-EF1-MSC2. ΔLNGFR was amplified with forward primer 5′-GCGGCTAGCGCCACCATGGGGGCAGGTGCCACCG-3′ and reverse primer 5′-GCGCTCGAGTCACCTCTTGAAGGCTATGTAG-3′ and ligated into the PmeI/SalI sites. GFP- and LNGFR-encoding sequences were cloned with Kozak sequences (Kozak, 1999). Plasmids were checked by sequencing (Medigenomix, Martinsried, Germany).
Nucleofection
CD4+ T cells were nucleofected with 5 μg of plasmid DNA at a cell density of 1 × 107 cells per 100 μl of Nucleofector solution (human T cell Nucleofector kit VPA-1002; Lonza) with the Amaxa Nucleofector device using program U-14 (Lonza). After nucleofection, cells were immediately transferred into prewarmed hTC culture medium (Lonza) supplemented with 2 mM glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% fetal calf serum (FCS; PAA Laboratories, Pasching, Austria). Transfection efficiencies were determined by flow cytometric analysis of GFP and LNGFR. Cells were cultured in 6-well plates in a humidified 37°C–5% CO2 incubator. Sixteen hours after nucleofection LNGFR-positive cells were enriched with anti-LNGFR microbeads (Miltenyi Biotec) and cultured with CD3/CD28 beads (Dynal Biotech, Oslo, Norway) at a ratio of 4:1 in the presence of recombinant human interleukin-2 (rHu IL-2) (Proleukin, 50 U/ml; Chiron/Novartis, Amsterdam, The Netherlands). Cells were restimulated with CD3/CD28 beads after each selection step.
SNARF-1 labeling
Proliferation of nucleofected T cells was measured by seminaphthorhodafluor (SNARF)-1 (carboxylic acid, acetate, succinimidyl ester) dilution as described previously (Magg and Albert, 2007). Briefly, a stock solution of 5 mM SNARF-1 (Invitrogen Molecular Probes, Eugene, OR) was prepared in dimethylsulfoxide (DMSO) and stored at −20°C. Freshly isolated T cells and nucleofected T cells were resuspended at a concentration of 5 × 106 cells/ml in PBS with 10% FCS. Cells were stained by rapidly mixing equal volumes of cell suspension with 25 μM SNARF-1 in PBS with 10% FCS and incubated at room temperature in the dark for 30 min. The labeled cells were washed three times with PBS–10% FCS before cultivation.
Flow cytometric analysis
T cells were stained for cell surface markers with phycoerythrin (PE)–anti-LNGFR (Miltenyi Biotec) and Tricolor (TC)–anti-CD4 (BD Biosciences, San Jose, CA). Apoptosis and cell death were quantified by Annexin V–PE and 7-amino-actinomycin D (7-AAD) staining (BD Biosciences). Flow cytometry was performed with a three-color FACScan (BD Biosciences). Data were analyzed with CellQuest (BD Biosciences) and WinMDI software (written by J. Trotter, BD Biosciences, San Diego, CA).
Western blot analysis
Whole cell extracts were prepared in Laemmli buffer 16 hr after transfection. Samples were separated on a sodium dodecyl sulfate (SDS)–10% polyacrylamide gel (Bio-Rad, Munich, Germany) and transferred to nitrocellulose membranes. Blocking was performed with 1% bovine serum albumin (BSA) in Tris-buffered saline. Membranes was probed with anti-turboGFP(d) antibody (Evrogen, Moscow, Russia) and anti-actin (Santa Cruz Biotechnology, Santa Cruz, CA). Secondary goat anti-mouse antibody and secondary goat anti-rabbit antibody were from Pierce Biotechnology (Rockford, IL).
[3H]Thymidine incorporation assay
A total of 5 × 104 T cells per well was incubated in replicates of four for the indicated time and pulsed with 0.5 μCi of [3H]thymidine per well for 8 hr. [3H]Thymidine incorporation was measured by liquid scintillation counting (Beckman Coulter, Fullerton, CA). Incorporated radioactivity is expressed as counts per minute.
Cytokine analysis
Culture supernatants were assayed for IL-4, IL-5, IL-10, IL-12, IL-13, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α, using the TH1/TH2 cytokine assay kit (Bio-Rad) according to the manufacturer's instructions and analyzed on the Bio-Plex 200 system (Bio-Rad).
Fluorescence microscopy
For fluorescence microscopy, 2.5 × 105 cells were stained with anti-LNGFR–PE (Miltenyi Biotec), centrifuged onto object slides at 200 × g for 3 min, and mounted in Aqua-Poly/Mount (Polysciences, Eppelheim, Germany). Laser-scanning microscopy was done with an Axiovert 135 microscope on an inverted stand, using the Plan-Apochromat 100/1.40 objective (Carl Zeiss MicroImaging, Göttingen, Germany). Counterstains were performed with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, Hamburg, Germany).
Analysis of genomic integration
Genomic DNA was isolated from 5 × 106 cells, using TRIzol reagent (Invitrogen, Paisley, UK) 35 days posttransfection. PCR was performed with Taq polymerase (New England BioLabs, Frankfurt, Germany), using 100 ng of genomic DNA with the following plasmid-specific primers: forward primer, 5′-CAGAGCTCGTTTAGTGAACC-3′ and reverse primer, 5′-GAGCCAGTACACGACATCAC-3′. The PCR program was set as follows: 5 min of denaturation at 94°C; 35 cycles of 40 sec at 94°C, 40 sec at 58°C, and 40 sec at 72°C; followed by 5 min at 72°C.
Southern blot analysis was performed with 10 μg of genomic DNA. Genomic DNA was digested with EcoRI, separated on a 0.8% Tris–acetate–EDTA (TAE)–agarose gel, and blotted on a nylon membrane (Schleicher & Schuell, Dassel, Germany). A 318-bp DNA probe specific for copGFP was synthesized with the primer pair 5′-TGATCGGCGACTTCAAGGTG-3′ and 5′-TTGAAGGCGTGCTGGTACTC-3′ from plasmid pCDH-CMV-GFP-EF1-LNGFR, using a PCR digoxigenin (DIG) probe synthesis kit (Roche, Mannheim, Germany). The probe was hybridized at 42°C overnight and detected with a DIG DNA labeling and detection kit (Roche).
Statistical analysis
Comparison of flow cytometry data and cytokine assays was done by two-tailed Student t test. Values of p less than 0.05 were considered to be significant.
Results
Efficient nucleofection and enrichment of primary human CD4+ T cells
The electroporation-based nucleofection method has been shown to be suitable even for “difficult-to-transfect” cells (Lai et al., 2003; Gresch et al., 2004). To evaluate transfection efficiency and viability of transfected primary human T cells, we used two different dual-expression vectors, each equipped with two different reporter and promoter systems. We expected higher expression efficiency with a transfection approach utilizing two separate promoters rather than a bicistronic expression strategy using an internal ribosomal entry site. The plasmid pCDH-CMV-LNGFR-EF1-GFP (pLNGFR/GFP) forces expression of GFP from the CMV promoter and ΔLNGFR from the EF1α promoter. In the plasmid pCDH-CMV-GFP-EF1-LNGFR (pGFP/LNGFR) the positions of the marker genes were mutually exchanged. The expression cassette of both plasmids is depicted in Fig. 1A.
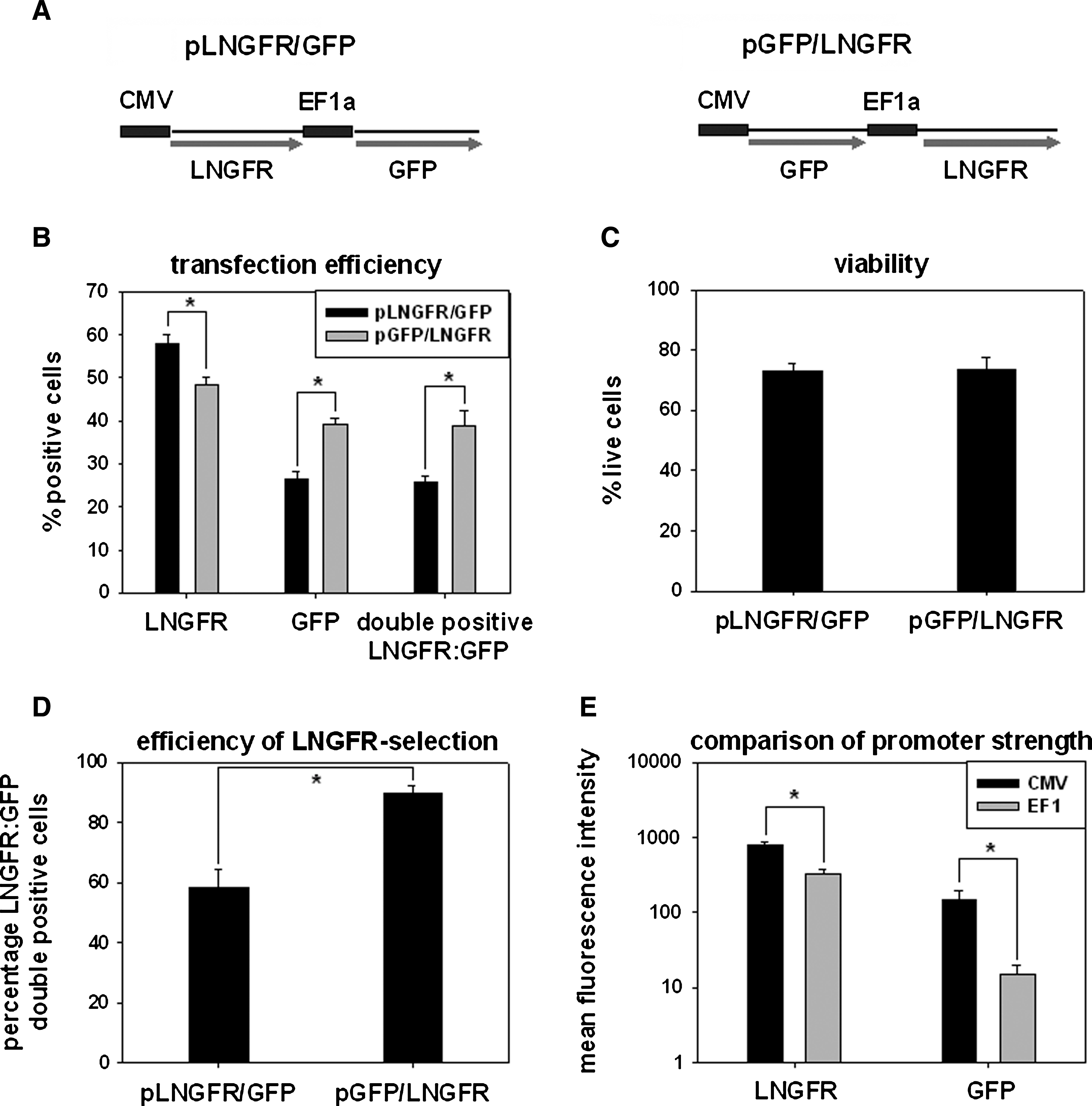
Nucleofection and enrichment of transfected primary human T cells. (
Transfection of primary human CD4+ T cells was performed with the Amaxa Nucleofector device. Transfection of the pLNGFR/GFP plasmid yielded 58.0 ± 2.0% LNGFR-positive cells, 26.4 ± 1.7% GFP-positive cells, and 25.6 ± 1.6% LNGFR/GFP double-positive cells. Although slightly fewer LNGFR-positive cells (48.5 ± 1.6%) were achieved with the pGFP/LNGFR plasmid, this resulted in a significantly higher proportion of LNGFR and GFP double-positive cells (38.4 ± 3.7) because of a higher frequency of GFP-positive cells (39.0 ± 1.4%) compared with pLNGFR/GFP (p = 0.0004; n = 5) (Fig. 1B). Cell viability was equal for both plasmids with 72.8 ± 1.9% viable cells for pLNGFR/GFP and 73.8 ± 1.9% for pGFP/LNGFR 16 hr posttransfection (p = 0.5; n = 5) (Fig. 1C). Transfected cells were enriched by immunomagnetic cell sorting with LNGFR microbeads 16 hr posttransfection. As expected, a higher percentage of LNGFR and GFP double-positive cells could be enriched with the pGFP/LNGFR expression plasmid, with 89.5 ± 2.5% double-positive cells compared with the pLNGFR/GFP expression plasmid with 58.4 ± 6.0% (n = 5, p = 0.00007) (Fig. 1D). The absolute expression levels of LNGFR and GFP were both dependent on the choice of promoter. In particular, 2.3-fold higher expression of LNGFR and 9.7-fold higher expression of GFP were obtained from the CMV promoter compared with their expression driven by the EF1α promoter (n = 5; p < 0.000001) (Fig. 1E). For this particular combination of promoter and reporter systems balanced expression of both reporters was achieved only when GFP was expressed from the CMV promoter and LNGFR from the EF1α promoter.
In summary, we could demonstrate simultaneous expression of two transgenes in primary human T cells with good efficiency by using a nonviral, dual-promoter plasmid transfection strategy. Because the highest percentage of LNGFR and GFP double-positive cells was obtained when expressing GFP under the control of the CMV promoter and LNGFR expression driven by the EF1α promoter, this expression plasmid, termed pGFP/LNGFR, was chosen for subsequent experiments.
Transient transgene expression in primary human T cells
To study effects of a particular ectopically expressed gene of interest it is necessary to achieve homogeneous gene expression in all cells. We therefore next examined whether positive selection with immunomagnetic beads would result in high-grade purification of the transfected cell population. Because we could not expect relevant genomic integration and stable transgene expression after nucleofection of resting T cells, we also examined the time course of reporter gene expression in cells stimulated after transfection.
Nucleofected primary human CD4+ T cells were positively selected 16 hr posttransfection with LNGFR-specific microbeads, resulting in a purity of 89.5 ± 2.5% double-positive cells (n = 5; Fig. 2A, third row). Consecutively LNGFR positively selected cells were stimulated with CD3/CD28 beads and cultured for 7 days. Because of delayed GFP expression the amount of double-positive cells slightly increased within 4 days to 96.3 ± 1.8% (n = 5; Fig. 2A, fourth row). Subsequently, cell proliferation led to a loss of expression of both reporter genes. Expression levels reached almost background level within 7 days (n = 5) (Fig. 2B).

Time course of GFP and LNGFR expression in nucleofected primary human CD4+ T cells. T cells were nucleofected with pGFP/LNGFR. (
Here we demonstrated that cotransfection of a surface antigen allows for immunomagnetic positive selection of highly purified CD4+ T cells, homogeneously but transiently transfected with a gene of interest.
Isolation of stably transfected primary human CD4+ T cells
Stable transfection is a prerequisite for long-term in vitro evaluation or possible clinical application of genetically modified T cells. We therefore next investigated whether our strategy would allow for the creation of stably transfected T cell lines.
Within 7 days of nucleofection of 107 cells the expression of both reporter proteins declined to background signal except for 0.5 ± 0.1% of the cells that remained double positive for LNGFR and GFP (n = 5; Fig. 3A). We next examined whether this remaining cell population represented stable transfectants. LNGFR/GFP double-positive cells were again enriched by magnetic cell sorting, yielding 21.9 ± 3.6% LNGFR/GFP double-positive cells with 4.9 × 105 ± 1.8 ×105 cells in total. After a second expansion period of 7 days with CD3/CD28 beads, 1 × 107 ± 2.4 × 106 cells were obtained with 1.8 ± 0.2% cells remaining LNGFR and GFP double positive (n = 5). After a third enrichment a total of 6.4 × 105 ±1.2 × 105 cells with a purity of 98.9 ± 0.6% could be harvested (n = 5; Fig. 3A). These T cells were cultured for 8 weeks after nucleofection and analyzed for LNGFR and GFP expression. Both marker genes remained at constantly high levels during the observation period (Fig. 3B). To confirm that these cells were indeed stable transfectants, genomic PCR analysis was performed with plasmid-specific oligonucleotides 35 days posttransfection. As internal reference for genomic DNA integrity and purity an intron-spanning β-actin-specific primer pair was used. A single PCR fragment was detectable in transfected T cells but not in nontransfected control T cells (Fig. 3C). We could further demonstrate by Southern blot analysis that the transgene was in fact integrated into the genome at multiple sites (Fig. 3D). To confirm that GFP protein was expressed strongly enough for immunodetection, whole cell lysates from stably transfected T cells 35 days posttransfection were prepared and separated by SDS–polyacrylamide gel electrophoresis (PAGE). A single 27-kDa protein band was detectable in transfected but not in nontransfected control cells (Fig. 3E). Using fluorescence microscopy we could further demonstrate the intracellular cytoplasmic location of GFP, whereas LNGFR expression was restricted to the cell surface (Fig. 3F).

Isolation of stably transfected primary human T cells. (
These findings implicate that nucleofection is suitable for the generation and purification of stably transfected primary human T cells. Stably transfected T cells can be maintained for several weeks without loss of gene expression.
Phenotypic characterization of transiently and stably transfected primary human T cells
We next addressed whether nucleofection would alter the normal T cell function of transiently and stably transfected T cells.
As shown in Fig. 4A nucleofected T cells exhibited a significant reduction in [3H]thymidine incorporation on T cell stimulation as compared with nontransfected control cells or stably transfected T cells, suggesting impaired DNA synthesis of freshly transfected T cells immediately after nucleofection (Fig. 4A). Freshly transfected T cells also exhibited significantly decreased secretion of IL-5, IL-10, IL-13 IFN-γ, and TNF-α but not IL-4 and IL-12 compared with nontransfected control cells, whereas stably transfected T cells showed a similar cytokine secretion pattern as nontransfected T cells cultured under identical conditions (Fig. 4B). To assess whether this was accompanied by a reduced capacity to proliferate, a fluorescence-based method for measuring cell division was used. Because of the spectroscopic incompatibility of GFP with the standard proliferation marker 5,6-carboxyfluorescein diacetate, succinimidyl ester (CFSE) we used the far-red fluorescent dye SNARF-1 as described previously (Magg and Albert, 2007). The SNARF-1 dilution profile of transiently and stably transfected T cells was compared with that of nontransfected T cells 5 days after stimulation with CD3/CD28 beads. The freshly transfected T cells displayed a slightly reduced proliferation capacity compared with control T cells (84.4 ± 3.0% proliferating cells vs. 95.0 ± 0.5%, n = 3; p = 0.004), whereas stably transfected T cells displayed a normal proliferative response compared with untransfected cells cultured under identical conditions (81.7 ± 2.2 vs. 79.9 ± 2.7%, n = 3; p = 0.51) (Fig. 4C). We then tested whether induction of apoptosis by the nucleofection procedure was the reason for these slightly decreased T cell function parameters. As shown in Fig. 4D, there was a significantly higher proportion of apoptotic (7-AAD/Annexin V double positive) cells on nucleofection during the first 16 hr of the culture period but not thereafter.

Cellular function of transiently and stably transfected primary human T cells. (
In summary, these data reveal a slight functional defect in freshly transfected T cells caused by excessive apoptosis induced by nucleofection. However, stably transfected cells showed normal T cell responses, implying that these cells could be candidates for the study of ectopically expressed genes or be applied therapeutically after genetic modification.
Discussion
Application of genetically modified T cells is a promising tool to treat malignancies, viral infections, genetic disorders, and autoimmune disease (Heslop et al., 1996; Pule et al., 2008). One limitation for the therapeutic use of genetically modified T cells is the low transfection efficiency of current techniques. The aim of this study was to establish an efficient and rapid method for enrichment of stably transfected primary human T cells. Here we describe for the first time an easy and reliable strategy for nonviral generation and purification of stably transfected primary human T cells exhibiting homogeneous long-term expression of a gene of interest.
In this study we used Amaxa nucleofection technology for nonviral DNA delivery (Gresch et al., 2004). As previously shown, several cell types can be successfully transfected by this technology (Hamm et al., 2002; Gresch et al., 2004; Schakowski et al., 2004). Using this system we created a nonviral gene transfer method for primary human T cells with satisfactory transfection efficiencies and high expression levels. Using a dual-expression vector, two different genes could be expressed by introducing a single plasmid facilitating expression of a gene of interest and a marker gene for positive selection of transfected cells. The C-terminal truncated low-affinity nerve growth factor receptor (ΔLNGFR) has been shown to be a bona fide marker gene enabling both fast detection by flow cytometry and efficient positive selection by immunomagnetic cell sorting (Phillips et al., 1996; Fehse et al., 1997). It has been previously shown that ΔLNGFR is a safe marker for T cells under conditions in which forced expression did not alter cellular function or survival (Bonini et al., 2003). Furthermore, ΔLNGFR is nonimmunogenic and has already been introduced in phase 1 clinical studies (Bordignon et al., 1995; Bonini et al., 1997). We used the well-established green fluorescent protein (GFP) as a second marker gene in this study to ensure easy quantification of transfection efficiencies and expression levels for a proof of principle. It should pose no significant challenge to exchange GFP or other genes of interest to be studied in human T cells.
In this study we used the CMV promoter and the EF1α promoter, which were both shown to be active in primary human T cells (Dardalhon et al., 2001). In contrast to the report from Dardalhon and colleagues we observed stronger expression driven by the CMV promoter as compared with the EF1α promoter. On selection with immunomagnetic beads ΔLNGFR-positive cells could be enriched to high purity, in line with previous studies that had utilized this surface molecule (Berger et al., 2003).
Tahvanainen and colleagues elegantly demonstrated by a similar approach the nonviral transfer of genes into primary human T cells. However, in their study gene expression was only transient and expression declined to almost background levels within days (Tahvanainen et al., 2006). Using our approach a small cell population with sustained expression of both marker genes was repeatedly observed. These obviously stably transfected cells made up about 0.5% of transfected cells. This relatively low ratio of stably transfected primary cells might have a negative impact on the T cell receptor repertoire of these cells. This might not necessarily be a disadvantage in therapeutic settings in which diverse polyclonal T cells are not of primary interest. Rather, for many therapeutic purposes it might be more desirable to generate T cells harboring either a transgenic T cell receptor of known specificity or to expand T cells with antigen-presenting cells (APCs) presenting a desired target antigen. The method presented here would allow for both T cell generation strategies.
Stably transfected cells could be concentrated by two subsequent enrichments with immunomagnetic beads. Stably transfected cells were maintained for at least 2 months without detectable loss of transgene expression, confirming the stability of the plasmid integration into cellular DNA. Thereby we could demonstrate an efficient and rapid way for generation of high-purity, gene-modified primary human T cells and also Jurkat cells (data not shown for Jurkat cells). Stable integration was verified by genomic PCR using plasmid-specific primers and by Southern hybridization, which successfully detected a plasmid-coding sequence integrated into genomic DNA in transfected but not in nontransfected control cells.
One major concern with gene-altered T cells is whether normal cellular functions were influenced when manipulating cells by introducing foreign DNA. Therefore we analyzed whether transfected T cells were able to respond appropriately on stimulation. Although the capacity of DNA synthesis, proliferation, and cytokine secretion is reduced shortly after transfection, we did not observe any differences in stably transfected T cells compared with nontransfected control cells.
Independent of the gene transfer method used, integration of plasmid DNA into the chromosome bears a risk for insertional mutagenesis. Indeed, in a clinical trial with genetically modified hematopoietic stem cells the induction of leukemia in patients has been reported (Hacein-Bey-Abina et al., 2003; Kohn et al., 2003). So far, however, no case of insertional mutagenesis has been reported when mature T cells were the target of gene correction, possibly due to their high grade of cellular differentiation.
In summary, this study provides proof of principle that human T cells can be altered to homogeneously and stably express a gene of interest via an easy, nonviral technique. This should enable further studies on T cell physiology and ultimately facilitate treatment approaches for diseases that are caused by defective gene function in T cells, such as some primary immunodeficiencies, or for diseases that require genetically designed T cell therapy.
Footnotes
Acknowledgments
This project was supported in part by a FoeFoLe grant (University of Munich, to T.M.) and by the Mehr LEBEN für krebskranke Kinder Bettina-Bräu-Stiftung (to M.H.A.).
Author Disclosure Statement
No competing financial interests exist for any of the authors.