
Abstract
In vivo gene transduction with adeno-associated virus (AAV)-based vectors depends on laborious procedures for the production of high-titer vector stocks. Purification steps for efficient clearance of impurities such as host cell proteins and empty vector particles are required to meet end-product specifications. Therefore, the development of alternative, realistic methods to facilitate a scalable virus recovery procedure is critical to promote in vivo investigations. However, the conventional purification procedure with resin-based packed-bed chromatography suffers from a number of limitations, including variations in pressure, slow pore diffusion, and large bed volumes. Here we have employed disposable high-performance anion- and cation-exchange membrane adsorbers to effectively purify recombinant viruses. As a result of isoelectric focusing analysis, the isoelectric point of empty particles was found to be significantly higher than that of packaged virions. Therefore, AAV vector purification with the membrane adsorbers was successful and allowed higher levels of gene transfer in vivo without remarkable signs of toxicity or inflammation. Electron microscopy of the AAV vector stocks obtained revealed highly purified virions with as few as 0.8% empty particles. Furthermore, the membrane adsorbers enabled recovery of AAV vectors in the transduced culture supernatant. Also, the ion-exchange enrichment of retroviral vectors bearing the amphotropic envelope was successful. This rapid and scalable viral purification protocol using disposable membrane adsorbers is particularly promising for in vivo experimentation and clinical investigations.
Introduction
In most situations, large-scale propagation of recombinant AAV uses transduction of HEK-293 cells and follows a streamlined concept of cell harvest, purification through CsCl density centrifugation, and concentration of the viral particle fraction. Conventionally, rAAV particles have been purified by rounds of isopycnic banding in CsCl density gradients (Merten et al., 2005). This method of purification is inconvenient because the development of the linear gradient takes greater than 24 hr per round in a standard ultracentrifuge. Consequently, the method of purification based on rounds of CsCl density centrifugation is not scalable and yields product of insufficient purity and transduction potency (Gao et al., 2000). Therefore, an alternative scalable purification procedure that is able to clear impurities including host cell proteins and empty particles is needed to meet end-product specifications.
Defined serum-free conditions have great conceptual advantages for biological safety and standardization of the production of clinical gene transfer vectors (Glimm et al., 1998). Although a conventional protocol to concentrate the retroviral vector can provide a high-titer stock (Tamura et al., 2001), accomplishment of the serum-free conditions has not yet been established. In addition, unlike the pseudotyped retroviral vectors bearing the vesicular stomatitis virus glycoprotein G (VSV-G), retroviral vectors bearing the amphotropic envelope protein cannot withstand the shearing forces imposed by ultracentrifugation. Efficient transduction with a high-titer serum-free vector stock can be realized only by using chromatographic approaches, making the virus-based vectors ideally suited for applications in clinical gene therapy.
Chromatography is widely used in the downstream purification steps of vector production (Sommer et al., 2003). However, the resin-based, packed-bed chromatography suffers from a number of limitations in practice. These are associated with pressure drops across the bed, slow pore diffusion, long cycle times, and difficulties in scaling up purification procedures (Knudsen et al., 2001). In this study, we have sought to develop an effective and convenient alternative for the removal of empty viral particles and enrichment of viral vector particles. We have adapted dual ion-exchange membrane chromatography to eliminate empty particles from cleared lysates containing recombinant viruses, using disposable ion-exchange membrane adsorbers. Although a previous report suggested the utility of the membrane-based technology in small-scale work (Duffy et al., 2005), scalable production as well as high-grade purification for in vivo experimentation were not distinctly demonstrated. In combination with our large-scale transduction technique using active gassing (Okada et al., 2005), we now have at hand a simple and highly efficient system to produce purified vector stocks. Furthermore, the membrane adsorbers enable effective recovery of the AAV vector in the supernatant of the transduced cell culture. Here, we present a high-throughput method for scalable purification of recombinant AAV or retrovirus, using disposable membrane adsorbers to establish a labor- and cost-effective purification system.
Materials and Methods
Cell culture
Propagation of vectors was based on transfection of human embryonic kidney-derived 293 (HEK-293) cells using a 10-tray cell factory (CF10; Nalge Nunc International, Rochester, NY) with a surface area of 6320 cm2 associated with an active gassing system, as described previously (Okada et al., 2005). Briefly, cells were cultured in Dulbecco's modified Eagle's medium and nutrient mixture F-12 (D-MEM/F-12; Invitrogen, Grand Island, NY) with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37°C in a 5% CO2 incubator. First, cells were plated at 6.5 × 107 cells per CF10 to achieve a monolayer of 20 to 40% confluency when cells initially attached to surface of the flask. The volume of medium used per flask was 1120 ml. Subsequently, cells were grown for 48–72 hr until reaching 70–90% confluence and were then transfected with appropriate plasmids. An aquarium pump (Nisso, Tokyo, Japan) was used to circulate the air through the CF10 with 5% CO2 with humidity in an incubator.
Canine skeletal myoblasts were isolated from a neonatal Beagle dog. Two grams of hind limb muscle was minced and dissociated with intermittent trituration to make a fine slurry. The mixture was filtrated with a BD Falcon cell strainer (100 μm; BD Biosciences, San Jose, CA) and was collected by centrifugation at 800 × g for 5 min. The cell pellet was resuspended with F-10 medium (Invitrogen) with 20% fetal calf serum (Sigma-Aldrich) and cultured on a collagen I-coated dish at 37°C in a 5% CO2 incubator. Three rounds of preplating were applied to diminish fibroblasts.
Construction and propagation of AAV vectors
A proviral plasmid harboring the EGFP gene (pAAV-EGFP) under the control of the CAG promoter, a modified chicken β-actin promoter with cytomegalovirus immediate-early (CMV-IE) enhancer, has been described previously (Okada et al., 2005). The rat interleukin (IL)-10 cDNA fragment was inserted into the EcoRI site of p3.3CAG-WPRE, which contains the CAG promoter and woodchuck posttranscriptional regulatory element (WPRE). The entire expression cassette was inserted between the AAV2-derived inverted terminal repeats (ITRs) in a pUC-based proviral plasmid, pAAVLacZ, to obtain pAAV1-IL-10.
Half the medium in the CF10 tissue culture flask was exchanged with fresh D-MEM/F-12 containing 10% fetal bovine serum (FBS), 1 hr before transfection of HEK-293 cells. Subsequently, the cells were cotransfected with 650 μg of each plasmid: proviral vector plasmid pAAV-EGFP, AAV-1 chimeric helper plasmid or AAV-8 chimeric helper plasmid, as well as adenoviral helper plasmid pAdeno, using calcium phosphate coprecipitation. Each plasmid was added to 112 ml of 300 mM CaCl2. This solution was gently added to the same volume of 2 × HBS (290 mM NaCl, 50 mM HEPES buffer, 1.5 mM Na2HPO4; pH 7.0) and gently inverted three times to form a uniform solution. This solution was immediately mixed with fresh D-MEM/F-12 containing 10% FBS to produce a homogeneous plasmid solution mixture. Subsequently, the medium in the culture flask was replaced with this plasmid solution mixture. At the end of a 6-hr incubation, the plasmid solution mixture in the culture flask was replaced with prewarmed fresh D-MEM/F-12 containing 2% FBS. Cell suspensions were collected 72 hr after transfection and centrifuged at 300 × g for 10 min. Cell pellets were resuspended in 30 ml of Tris-buffered saline (TBS: 100 mM Tris-HCl [pH 8.0], 150 mM NaCl). Recombinant AAV was harvested by five cycles of freeze–thawing of the resuspended pellet. The crude viral lysate was initially concentrated by a brief two-tier CsCl gradient centrifugation for 3 hr (Okada et al., 2002) and then the vector fraction was dialyzed in MHA buffer (3.3 mM morpholineethanesulfonic acid [MES], 3.3 mM HEPES [pH 6.5, 7.0, 7.5, or 8.0], 3.3 mM sodium acetate).
In parallel with processing the cell pellets, the culture supernatant sample was also processed for the dual ion-exchange procedure by centrifugation and filtration. The culture supernatant fluid, 72 hr after transduction, was sampled and clarified with an appropriate amount of activated charcoal (Wako Pure Chemical Industries, Osaka, Japan). Insoluble debris was removed by centrifugation at 3000 × g for 15 min and filtration. The elucidated culture supernatant was desalted as well as enriched with a hollow fiber cross-flow membrane (100,000 nominal molecular weight cutoff [NMWC]; GE Healthcare, Piscataway, NJ). The sample was concentrated by a brief two-tier CsCl gradient centrifugation for 3 hr and then the vector fraction was dialyzed in MHA buffer.
Construction and propagation of retroviral vectors
Luciferase cDNA was cloned from the pGL3-Basic vector (Promega, Madison, WI), and inserted into pGCDNsap (Suzuki et al., 2002) to obtain pDNLuc. To generate a retroviral vector bearing the amphotropic envelope protein or a VSV-G-pseudotyped retroviral vector on a large scale, HEK-293 cells were transduced with 650 μg of pDNLuc, 325 μg of pGag-pol (Ory et al., 1996), and 325 μg of pEnv (Okada et al., 2004) or pVSV-G (Ory et al., 1996) in the CF10 culture flask. Culture supernatant was collected 48 hr after transduction and centrifuged at 300 × g for 3 min to remove cell debris. To enrich the recombinant retrovirus, vector-containing supernatant was centrifuged at 50,000 × g for 2 hr or at 6,000 × g for 16 hr at 4°C. Alternatively, the vector was concentrated and purified by ion-exchange chromatography.
Dual ion-exchange chromatography
Chromatography was performed with an ÄKTAexplorer 10S fast protein liquid chromatography (FPLC) system (GE Healthcare Life Sciences) equipped with a 50-ml Superloop (GE Healthcare Life Sciences). A dialyzed vector-containing fraction was loaded onto a cation-exchange membrane with a column volume of 0.18 ml (Acrodisc unit with Mustang S membrane; Pall, East Hills, NY) equilibrated with MHA buffer. After loading at a rate of 3 ml/min, the membrane was washed with 10 column volumes of MHA buffer. Bound samples on the Mustang S membrane was eluted over a 50-column volume span with a 0–2 M linear NaCl gradient in MHA buffer and 1-ml fractions were collected. The eluted sample from the Mustang S membrane was concentrated with a Macrosep 100K Omega filter (Pall) and used for electron microscopic assessment.
The flow-through sample that passed through the Mustang S membrane was diluted with 10 volumes of MHA buffer and further loaded at a rate of 3 ml/min onto an anion-exchange membrane with a bed volume of 0.18 ml (Acrodisc unit with Mustang Q membrane; Pall) equilibrated with MHA buffer. The membrane was then washed with 10 column volumes of MHA buffer. Bound virus on the Mustang Q membrane was eluted over a 50-column volume span with a 0–2 M linear NaCl gradient in MHA buffer and 1-ml fractions were collected. Recombinant rAAV particle number was determined by quantitative polymerase chain reaction (Q-PCR) of DNase I-treated stocks with plasmid standards.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and staining
A peak vector-containing fraction from the anion-exchange membrane (typically fraction 3) was desalted with a Macrosep 100K Omega filter and separated in 0.9-mm-thick 4–20% gradient polyacrylamide gels (Daiichi Pure Chemicals, Tokyo, Japan) with a sodium dodecyl sulfate (SDS) running buffer. Negative staining of the gel was performed with a Snow White kit (Mo Bi Tec, Göttingen, Germany) as per the manufacturer's instructions. Purity of viral band (VP1, VP2, and VP3) was analyzed with NIH Image (version 1.63) software.
Electron microscopy
Samples (10 μl) were spotted onto a carbon-stabilized copper specimen grid and incubated for 1 min. Excess sample was removed with a small piece of filter paper. The samples were then negatively stained for 1 min with 1% uranyl acetate solution. After removal of excess uranyl acetate, the air-dried grids were viewed with a JEM 2000EX transmission electron microscope (JEOL, Tokyo, Japan) at an accelerating voltage of 100 kV.
Isoelectric focusing
Isoelectric focusing (IEF) is an electrophoresis molecular diagnostic operation that separates protein in a pH gradient according to their isoelectric points (pI). Samples containing empty particles as well as fully packaged virions certified by electron microscopy were desalted with Amicon-30 (Millipore, Billerica, MA), and evaluated by IEF. Small-format Novex pH 3–10 IEF gels (EC6655A; Invitrogen, Carlsbad, CA) were used in a Novex XCell SureLock mini-cell (Invitrogen) to separate samples and Serva IEF protein markers 3–10 (39212-01; Invitrogen) as per the manufacturer's instructions.
Transduction experiments
To confirm transgene expression by the propagated vector in vivo, 3-week-old male Wistar rats were injected via the anterior tibial muscle with either CsCl-purified vectors (control, n = 12) or ion-exchange membrane-purified AAV1-IL-10 (CsCl + Mustang S/Q [MtgS/Q], n = 8) at 6 × 1010 genome copies per animal. Eight weeks after injection, serum IL-10 concentration was estimated by enzyme-linked immunosorbent assay (ELISA) (GE Healthcare Life Sciences; R&D Systems, Minneapolis, MN). Histological analysis of the muscle injection site was performed to examine the tissue for signs of toxicity or inflammation. To analyze the indication in muscle, which is susceptible to virus-mediated inflammation, we used mdx mice, a mouse model of Duchenne muscular dystrophy (DMD). Dystrophic muscles injected with the AAV vector reveal drastic immune response, which might be associated with the dystrophin-deficient sarcolemma of muscle fibers (Yuasa et al., 2002). To avoid the effect of transgene, viruses were inactivated by ultraviolet irradiation for 30 min before injection. Male mdx mice were injected with either PBS, CsCl-purified particles, or ion-exchange membrane-purified particles of AAV1-IL-10 at 1 × 1011 genome copies per animal via the tibialis anterior muscle at 2 weeks old (n = 2, each), when the pathology is indistinct. Two weeks after injection, the mice were killed and muscle samples were stained with hematoxylin and eosin (H&E) to analyze the pathology.
Canine myoblasts were transduced with rAAV-EGFP at 1 × 105 or 3 × 105 genome copies per cell to observe transgene expression under a fluorescence microscope (IX-70; Olympus, Tokyo, Japan) 3 days after infection. U251MG human glioma cells were transduced with luciferase-expressing recombinant retroviruses to evaluate the effect of the various purification procedures. At 96 hr after transduction, luciferase expression in the cells was analyzed with the Bright-Glo luciferase assay system (Promega).
Results
Effect of pH on rAAV recovery
The ion-exchange procedure as a downstream purification was compatible with the initial CsCl density centrifugation when vector-containing fractions were desalted by dialysis. rAAV type 1 (rAAV-1) was successfully recovered by ion-exchange chromatography and pH was an effective parameter to control viral particle affinity for the membrane (Table 1). Optimal recovery of vector genomes by the cation-exchange procedure was achieved at pH 6.5, whereas favorable recovery by the anion-exchange procedure was observed at pH 7.5–8.0.
Abbreviation: GC, genome copies.
Particles in the flow-through sample from the cation-exchange procedure were bound and eluted during anion exchange.
Characterization of rAAV purified by dual ion-exchange chromatography
rAAV-1-containing samples collected by a quick two-tier CsCl gradient centrifugation were dialyzed and applied to a cation-exchange membrane, to capture the impurities including particles. Figure 1a demonstrates the chromatogram of the cation-exchange procedure. Subsequently, the unbound flow-through sample in the cation-exchange procedure was applied to an anion-exchange membrane. The chromatogram in Fig. 1b demonstrates the resolution and dynamic binding capacity of the anion-exchange membrane. The eluted samples were desalted and concentrated by ultrafiltration. Elution profiles of the samples were stably maintained over the repeated purification procedures, although the fraction number of the vector-containing sample can be altered when the elution speed or gradient conditions are modified.

Elution profile of rAAV-1, using the dual ion-exchange method. (
Electron microscopic analysis of the samples collected from the two-tier CsCl centrifugation revealed virions with more than 6% empty particles (Fig. 2a; 91 empty particles out of 1350 virion particles, 6.7%). Interestingly, analysis of the sample eluted from the cation-exchange membrane revealed that the vast majority of the sample was empty particles (Fig. 2b; 694 of 713 particles, 97.3%). On the other hand, the unbound flow-through sample in the cation-exchange procedure was sequentially applied to an anion-exchange procedure, as dual ion-exchange purification. The samples obtained after the dual ion-exchange purification revealed highly purified virions with as few as 0.8% empty particles (Fig. 2c; 26 of 3365 particles). The analysis was repeated with similar results.
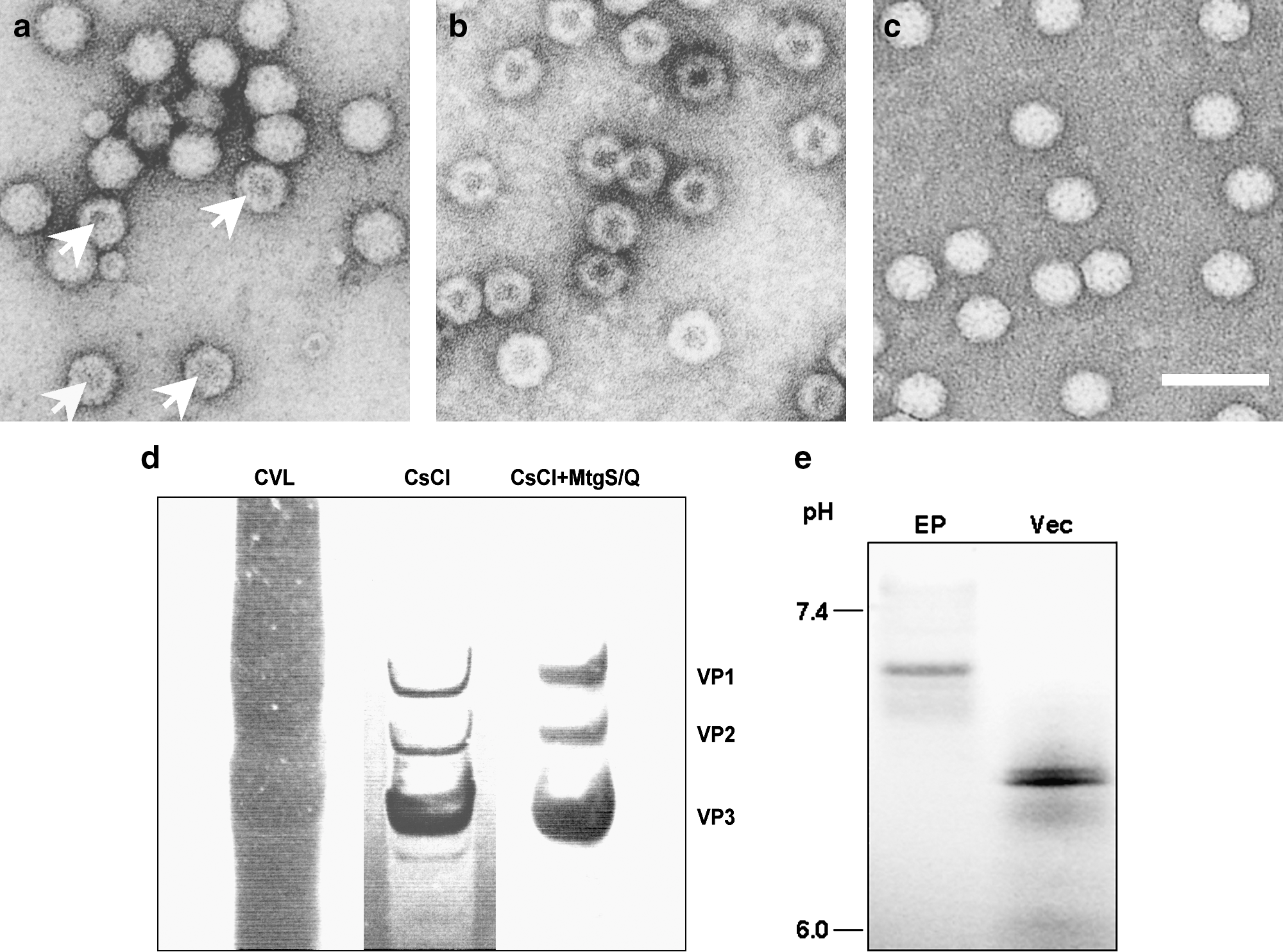
Electron microscopic assessment of empty particle contamination by dual ion-exchange chromatography. (
When the viral lysate was purified using dual ion-exchange adsorptive membranes after CsCl centrifugation, vector stocks with greater than 90% purity, as judged by SDS–PAGE analysis, were produced (Fig. 2d).
To estimate the difference in net charge of empty particles and packaged virions, IEF was performed. As the samples move through the gradient, they encounter a point where the pH is equal to their pI and they stop migrating. The pI of the empty particles was significantly higher than that of packaged virions (Fig. 2e).
Effect of purification on transduction efficiency
To confirm transgene expression by the propagated vector in vivo, 3-week-old male Wistar rats were injected via the anterior tibial muscle with either CsCl-purified vectors (n = 12) or ion-exchange membrane-purified AAV1-IL-10 (n = 8) at 6 × 1010 genome copies per animal. Serum IL-10 concentrations in animals transduced with the ion-exchange membrane-purified vectors were significantly higher than in the controls transduced with CsCl-purified vectors (519 ± 261 vs. 148 ± 83 pg/ml; p < 0.001) (Fig. 3a).

Improved transduction of muscle. (
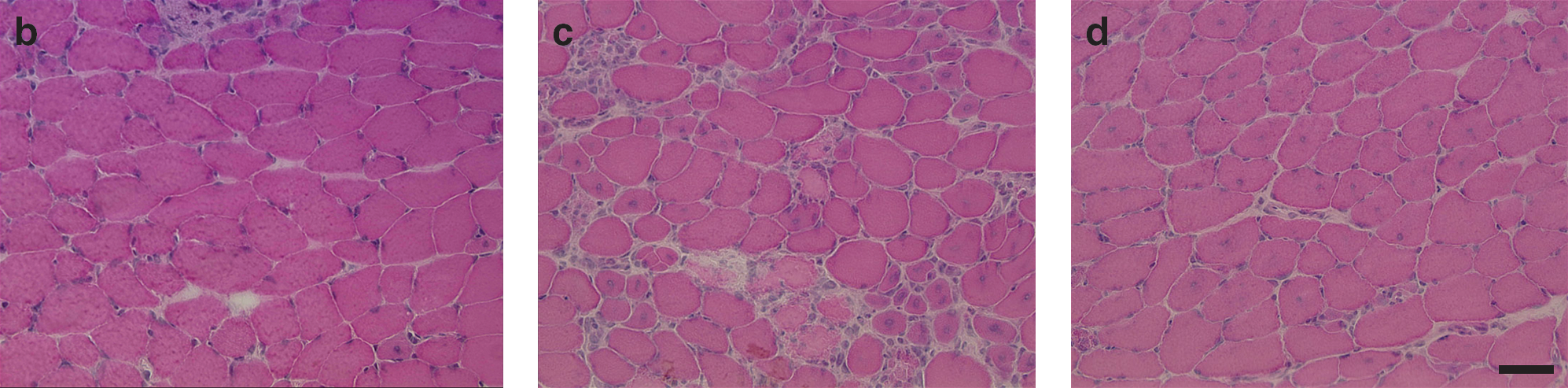
Injection of the ion-exchange membrane-purified vector particles into dystrophic muscles verified minimal inflammation. To analyze the effect on the histology of the tissue that is susceptible to virus-mediated inflammation, we used mdx mice, a mouse model of DMD. The tibialis anterior muscle of control mice injected with PBS showed scarce inflammatory cell infiltration, along with myofibers with central nuclei (Fig. 3b). mdx mice injected with CsCl-purified particles demonstrated extensive cellular infiltration with polymorphonuclear and mononuclear cells at the site of injection (Fig. 3c). In contrast, mice injected with the ion-exchange membrane-purified particles revealed manifestation similar to that of the PBS-treated control at 4 weeks of age (Fig. 3d).
Scalable vector preparations with dual ion-exchange chromatography
The average recovery of purified recombinant AAV-1 from four different crude virus lysates was 17.4% (data not shown). Although vector yield was dependent on the transgene and construct, recovery up to 2.0 × 1014 vector particles per dual Acrodiscs was achieved. Interestingly, we found that the yield from the supernatant was significantly higher than that from the cell pellet (Table 2). The amount of DNase-resistant particles in the supernatant was 7.3-fold higher than that obtained from the cell pellet (n = 7, p < 0.001). Furthermore, the ion-exchange protocol using adsorptive membranes was tried to realize effective purification. The bed volume of the adsorptive membrane was only 0.18 ml and this is much less than that of a conventional resin-based column with identical protein capture capacity. Therefore, every step, including capture, wash, and elution procedure, was performed quickly. Consequently, the membrane adsorbers enabled the efficient recovery of rAAV-8 in the 500-ml supernatant of the transduced cells culture (Table 3). To confirm the transduction activity, canine myoblasts were infected with the rAAV8-EGFP produced (Fig. 4). Efficiency of transduction of myoblasts with vectors derived from the culture supernatant was equivalent to that from the cells.
Ratio of particle numbers in supernatant to those in cell pellet.
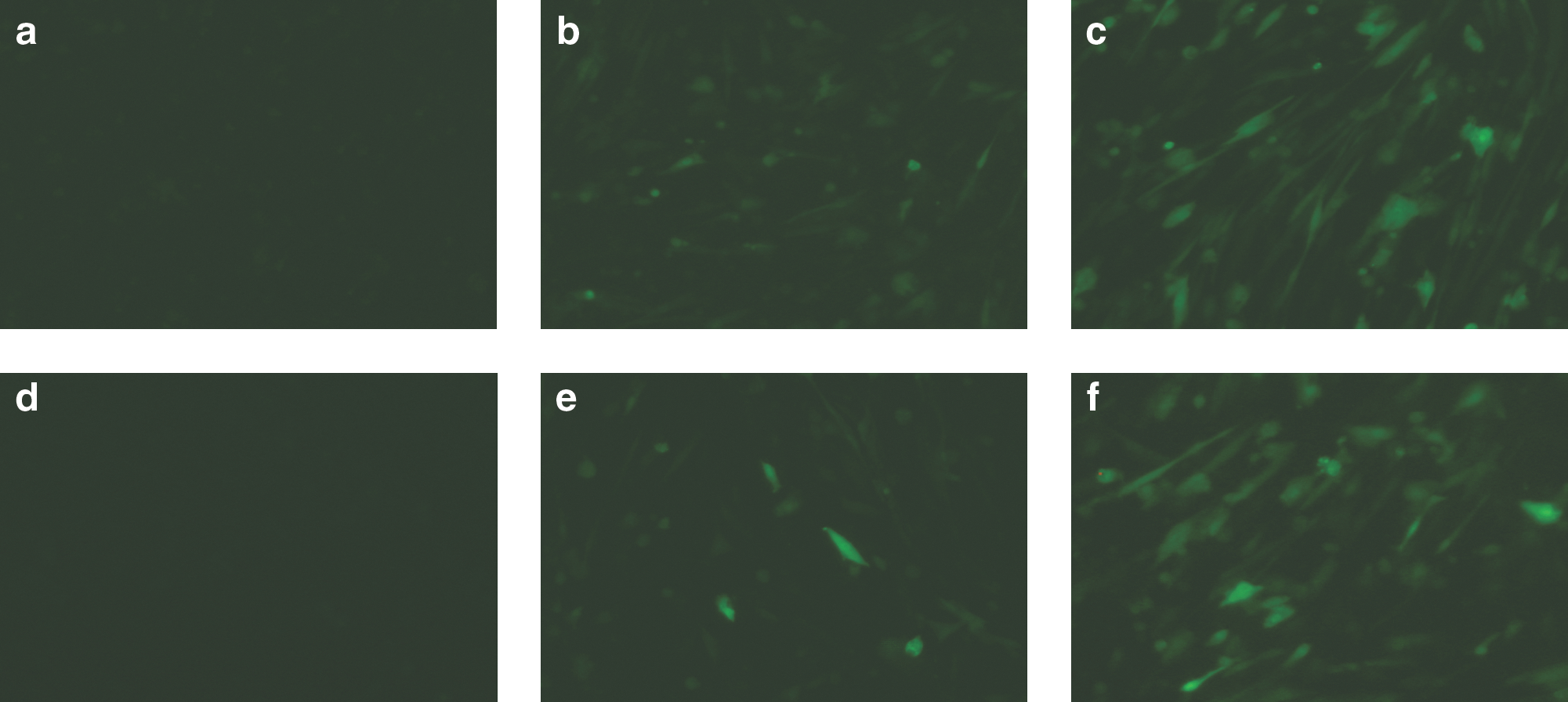
Myoblast transduction with rAAV8 purified from cell pellet or culture supernatant. rAAV8-EGFP was purified from the (
Particles loaded, vector genomes applied to the dual-ion exchange procedure.
Data are from cation exchange at pH 6.5 and anion step at pH 8.0.
U251MG human glioma cells were transduced with the luciferase-expressing recombinant retroviruses obtained from the various purification procedures (see Supplementary Table 1 at
Discussion
In this study, we have described an effective and scalable method for the purification and concentration of rAAV particles as well as amphotropic retroviral vectors. This straightforward method uses cation-exchange followed by anion-exchange chromatography to obtain purified rAAV particles. The AAV vector preparation with dual ion-exchange chromatography allowed higher levels of gene transfer in vivo without remarkable signs of toxicity or inflammation. Furthermore, this scalable strategy enabled the recovery of rAAV particles from the transduction culture supernatant, which is normally discarded in the course of the purification procedure. Also, the chromatographic method of concentrating amphotropic retroviral vectors successfully generated serum-free viral stocks with efficient recovery.
Membrane adsorbers provide an attractive alternative to traditional bead-based chromatography columns used to remove impurities in various applications (Phillips et al., 2005). Bead ligands in the packed columns are reached only by long-distance diffusion through pores. Therefore, the purification procedure with resin-based packed-bed chromatography is lengthy, including equilibration, sample application, washing, and elution. In addition, contamination with the distinct vectors will often recur if cost-effective disposable material has not been prepared. In contrast, binding sites in the membrane adsorbers are exposed to the molecules within short diffusion distances. This improved accessibility to the adsorber material makes vector purification processing much easier than with packed-bed chromatography and minimizes the essential bed volume. These characteristics of the disposable ion-exchange membrane save substantial time as well as viral viability. AAV vector purification with membrane adsorbers was effective and allowed higher levels of gene transfer in vivo without any signs of toxicity or inflammation. Electron microscopy of the AAV vector stocks obtained revealed highly purified virions with as few as 0.8% empty particles.
IEF is an electrophoresis molecular diagnostic operation that separates protein in a pH gradient according to their pI. Because of differences in pI, different proteins will focus at different points in the gradient. The pI is the specific pH at which the net charge of the protein is zero. The net charge of a virus is the sum of all the negative and positive charges of its amino acid side chains and amino and carboxyl termini along with the packaged DNA in the virion. In this study, we have shown that the pI of empty particles was significantly higher than that of packaged virions. Because proteins are positively charged at pH values below their pI and negatively charged at pH values above their pI, the net positive charge of empty particles would be higher than that of packaged virions at a constant pH below the pI of empty particles (see Supplementary Figure 1 at
Interestingly, we found that the yield from the supernatant was significantly higher than that from the cell pellet. This is probably due to the transduction event in that crumpled transduced cells released a considerable amount of rAAV particles into the culture medium. Therefore, it is practically and theoretically reasonable to use the supernatant of the transduction culture for rAAV production. However, conventional ion-exchange chromatography, using resin bead-based columns with a huge bed volume size, is a time-consuming procedure. In this respect, the protocol described here, in which small membrane absorbers are used, is an effective and quicker technique.
The method described herein also has several other attractive features. First, purification of rAAV particles with disposable membrane obviates concerns about vector contamination by other infectious agents potentially trapped in an ion-exchange column used for multiple purification procedures. Although a brief centrifugation on CsCl density gradients is used as an initial step, the method reported here does not require high-speed centrifugation over 24 hr, and avoids rounds of isopycnic banding steps to collect vector-containing fractions. In addition, the vector binding and elution properties achieved with the buffers and membrane adsorptive chromatography system produced high-titer vector. Dual ion-exchange chromatography with a brief centrifugation on two-tier CsCl density gradients can effectively avoid destruction of viral particles, which may contribute to the improved transduction efficiency in vivo (Kaludov et al., 2002). Future improvements of this method could be made by devising a method to eliminate the initial concentration step by CsCl density gradient. Potentially, the use of detergents, optimized ultrafiltration, or an affinity chromatography could remove a larger percentage of the impurities in the crude lysate so that we may be able to skip the initial two-tier CsCl density gradients.
As a result of our purification procedure, pure stocks with less than 1% empty particles were successfully produced. Although a previous protocol using conventional resin-based anion-exchange chromatography showed reduced contamination by empty particles in the rAAV-1 stock, empty particle contamination of samples was approximately 5% (Urabe et al., 2006). In our study here, elimination of the various impurities might be associated with the better transduction efficiency and minimal inflammatory reaction by dual ion-exchange chromatography compared with CsCl centrifugation. In addition to the effective reduction of empty particles, this protocol also successfully avoided lengthy exposure to CsCl. The conventional method of purification based on rounds of CsCl density centrifugation yields vectors with reduced transduction activity (Gao et al., 2000).
Unlike pseudotyped retroviral vectors bearing the VSV-G protein, retroviral vectors bearing the amphotropic envelope protein cannot withstand the shearing forces of ultracentrifugation. Therefore, the utility of the concentrated amphotropic retroviral vector is limited in the in vivo transduction experiment, although it is less toxic compared with the VSV-G-pseudotyped retroviral vector. Besides, suitable transduction medium usually varies with the target cells, although retroviral vector is collected as a vector-containing soup with medium used for 293 cells transduction. However, selection of the medium for 293 cell transfection frequently conflicts with the culture conditions for hematopoietic cells, although it is difficult to replace the suspension medium after harvest of the vector. In contrast, the quick ion-exchange chromatography resulted in good recovery of biologically active recombinant viruses with the amphotropic envelope protein. Elimination of the various impurities might be associated with better recovery of vectors by dual ion-exchange chromatography compared with anion exchange alone.
The production and purification procedures described in this report will expedite gene therapy research by providing a simple, rapid, and effective method for the generation of highly purified vectors. In addition, these procedures may potentially be applied to the production and purification of other vector systems. This closed viral production protocol using disposable membranes is particularly promising, as it easily interfaces with standard FPLC systems as well as syringe pumps. Moreover, the disposable material represents an advantage to meet GMP specifications. This system will provide the basis for further in vivo experiments and future clinical investigations.
Footnotes
Acknowledgments
The authors thank Dr. James M. Wilson for providing chimeric helper plasmids for rAAV1 (p5E18RXCI) and rAAV8 (p5E18-VD2/8). The authors thank Avigen (Alameda, CA) for providing pAAV-LacZ and pAdeno, Dr. M. Onodera (Tsukuba University, Japan) for pGCDNsap, Dr. T. Hope (University of Illinois, Chicago, IL) for pBS II SK+WPRE-B11, and Dr. J.I. Miyazaki (Osaka University, Japan) for pCAGGS. The authors thank Dr. Mikiharu Yoshida for helpful discussion, and also Ms. Miyoko Mitsu and Ms. Kiyomi Aoki for encouragement and technical support. This study was supported in part by (1) grants from the Ministry of Health, Labor, and Welfare of Japan, (2) Grants-in-Aid for Scientific Research, (3) a grant from the 21st Century COE program, and (4) a “High-Tech Research Center” Project for Private Universities matching fund subsidy, from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author Disclosure Statement
No competing financial interests exist for all authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.