
Abstract
One of the main objectives of cancer therapy is to enhance the effectiveness of the drug by concentrating it at the target site and to minimize the undesired side effects to nontarget cells. We have previously constructed a fusion protein, Lodavin, consisting of avidin and the endocytotic part of the low-density lipoprotein receptor, and demonstrated its applicability to transient drug targeting in vivo. In this study we produced a lentiviral vector expressing this fusion protein and evaluated its safety and efficacy. The results showed that lentivirus-mediated gene transfer led to long-term avidin fusion protein expression on glioma cells and that the receptor was able to bind biotinylated compounds. Repeated administration was proven feasible and the optimal time frame(s) for administration of biotinylated therapeutic and/or imaging compounds was elucidated. Intravenous or intracranial injection of the virus into BDIX rats led to the production of antibodies against transgene (avidin), but repeated administration of the vector was unable to boost this effect. Neutralizing antibodies against the lentivirus were also detected. Furthermore, we showed that the anti-avidin antibodies did not significantly affect the ligand-binding capacity of the avidin fusion protein. The therapeutic efficacy of avidin fusion protein in tumor treatment was tested in vitro with biotinylated and nonbiotinylated nanoparticles loaded with paclitaxel. In vivo applicability of lentivirus was studied in the BDIX rat glioma model, in which high receptor expression was detected in the tumor area. The lentivirus-mediated delivery of the avidin fusion protein thus represents a potential approach for the repeated targeting of cytotoxic compounds to cancer cells.
Introduction
A fusion protein consisting of avidin and the endocytotic part of the low-density lipoprotein receptor (LDLR) has previously been constructed by our group in order to achieve pretargeting of biotinylated molecules after local gene transfer (Lehtolainen et al., 2003). It takes advantage of the extremely high affinity of avidin for biotin (K d, 10–15 M) (Green, 1990). Semliki Forest virus (SFV) vector was chosen for preliminary studies because of its efficient gene transfer capacity and rapid onset of transgene expression. However, there are challenges in using SFV, such as short expression time (about 3 days) and potential neurotoxicity (Frolov et al., 1996; Rheme et al., 2005). Driven by the need for longer and safer transgene expression, a lentiviral vector expressing the avidin fusion protein was generated. As integrating vectors, lentiviruses are ideal for achieving long-term transgene expression (Naldini et al., 1996). They are nonlytic and considered to be less immunogenic than other vectors (Bessis et al., 2004). More importantly, long-term expression of avidin fusion protein would enable multiple administrations of biotinylated therapeutic agents, thus enhancing their therapeutic effect.
The aim of this study was to evaluate the safety and efficiency of the lentivirus-mediated expression of avidin fusion protein, to determine the ratio of total to surface expression of avidin in glioma cells, and to characterize its biotin-binding capability after single or repeated administration of biotinylated molecules. Previous studies have shown that the immune response can lead to elimination of vector, transduced cells, or therapeutic protein, thus decreasing the effect and duration of treatment (Bessis et al., 2004; Raty et al., 2008). The antibody response against lentiviral vector and the avidin fusion protein was studied in vivo with intracranial and intravenous delivery. Readministration of the lentiviral vector was studied to determine the influence and strength of the immune response after rechallenge. Finally, the efficacy of treatment was evaluated in vitro and expression of the avidin fusion protein in vivo was demonstrated. In conclusion, we demonstrate that lentivirus-mediated delivery of avidin fusion protein to tumor cells is an attractive approach for targeted cancer therapy.
Materials and Methods
Lentivirus expressing avidin fusion protein
A gene encoding LDLR–avidin fusion protein, Lodavin (Lehtolainen et al., 2003), was cloned into a third-generation self-inactivating lentivirus transfer plasmid under the control of the CAG promoter (chicken β-actin promoter with the cytomegalovirus [CMV] immediate-early enhancer upstream and a splicing acceptor site from the β-globin gene downstream). The constructs were verified by sequencing. Avidin fusion protein-expressing lentiviruses were prepared in triple flasks by a calcium phosphate transfection method in 293T cells as described previously (Follenzi and Naldini, 2002) and concentrated by ultracentrifugation. Testing of replication-competent lentiviruses (RCLs) was done by the p24-based assay method (Sastry et al., 2003) from cell culture supernatants (HeLa cells) up to 4 weeks.
Titer determination
The concentration of lentiviral capsid protein p24 (pg/ml) was determined with a p24 enzyme-linked immunosorbent assay (ELISA) kit (PerkinElmer, Waltham, MA). Vector particle quantification (total particles), determined with an ELISA test to the lentiviral capsid protein p24, was based on the estimation that a human immunodeficiency virus (HIV) core particle is composed of 2000 capsid proteins (Farson et al., 2001) and thus 1 pg of p24 represents 12,500 viral particles. The p24 amounts were converted to transducing units per milliliter by multiplying them with a conversion factor obtained by dividing the green fluorescent protein (GFP) control virus fluorescence-activated cell-sorting (FACS) titer (TU/ml, determined in HeLa cells) by its p24 amount, assayed simultaneously with the vector sample.
RNA titer was determined by a one-step reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) method. Briefly, RNA was isolated with a QIAamp MinElute virus spin kit (Qiagen, Valencia, CA) from 40 μl of concentrated vector, treated with TURBO DNA-free DNase (Applied Biosystems, Foster City, CA), and inactivated with DNase inactivation reagent. One-step RT-PCR was performed with an AgPath-ID one-step RT-PCR kit (Applied Biosystems), using the following primer/TaqMan MGB (minor groove-binding) probe set: Fw, 5′-GCTGCTGCGGTCAAGTGTTA-3′; Rev, 5′-TGCCGACCCTGGTAGCTTT-3′; probe, 5′-TGACATTGGTGATGACTG-3′. A plasmid standard was used to calculate RNA copy numbers from each sample.
The functional viral titer (TU/ml) was analyzed by transducing human glioma U-87 MG (American Type Culture Collection [ATCC], Manassas, VA) or rat glioma BT4C (Tyynela et al., 2002) cells (seeded 1 day previously on a 6-well plate) with various dilutions of the viral preparation 3 and 10 days (U-87 MG) later and analyzing avidin-positive cells by flow cytometry (see the next section) or by determining the total number of vector DNAs in transduced U-87 MG cells with an UltraRapid lentiviral titer kit (System Biosciences, Mountain View, CA). The DNA titers were corrected by subtracting out the ampicillin resistance (AmpR) plasmid background measured by qPCR. The AmpR qPCR was carried out with TaqMan gene expression master mix (Applied Biosystems) and the following primer/TaqMan MGB probe set: Fw, 5′-GTGTCGCCCTTATTCCCTTTT-3′; Rev, 5′-GCGTTTCTGGGTGAGCAAA-3′; probe, 5′-TGCGGCATTTTGCCT-3′. A plasmid standard was used to calculate plasmid copy numbers from each sample.
Avidin fusion protein expression in vitro
For viral titering the cells were dissociated with cell dissociation buffer (Invitrogen, Carlsbad, CA) and fixed with Cytofix/Cytoperm solution (BD Biosciences, San Jose, CA). Cells were washed with 1× BD Perm/Wash buffer (BD Biosciences) and blocked with 1× BD Perm/Wash buffer containing 0.1× FcR blocking reagent (Miltenyi Biotec, Bergisch Gladbach, Germany). Anti-avidin–fluorescein (diluted 1:35; Vector Laboratories, Burlingame, CA) was used to detect avidin. All the incubations were done on ice. Stained samples were suspended in phosphate-buffered saline (PBS) and analyzed by flow cytometry (BD FACSCanto II; BD Biosciences). Titers were calculated from flow cytometric results as described (Follenzi and Naldini, 2002), showing ≤10% (Logan et al., 2004).
To analyze total and surface expression and ligand binding, U-87 MG cells were transduced at a viral dilution of 1:200. To analyze total expression (tE analysis) from permeabilized cells the staining was performed as described previously except that goat anti-avidin D (diluted 1:40; Vector Laboratories) was used followed by Alexa Fluor 647-conjugated donkey anti-goat (1:200; Invitrogen) secondary antibody staining. For the staining of surface expression (S) cells were fixed with Cytofix solution (BD Biosciences) and stained with the same primary and secondary antibodies diluted in 1× BD stain buffer. FACS analysis was carried out in a buffer containing 0.5% bovine serum albumin (BSA) and 10 mM HEPES. The mean fluorescence index was calculated by multiplying the percentage of positive cells by the mean fluorescence.
Expression was also studied by fluorescence microscopy. Cells were seeded onto coverslips and stained with anti-avidin–fluorescein, according to the same principle as described previously. Mowiol was used for embedding. Cells were imaged by fluorescence microscopy (BX-81; Olympus America, Center Valley, PA).
To analyze the duration of avidin fusion protein total expression in BT4C and U-87 MG cells, virus dilutions of 1:70–1:100 were used and stainings were performed as described previously. For fluorescence-activated cell sorting of the positive cells, the cells were stained with the primary and secondary antibodies diluted in 1% BSA, 10 mM HEPES in PBS.
To study pseudotransduction, U-87 MG and BT4C cells were transduced with a 1:100 dilution of the viral stock in growth medium containing 1 μM 3′-azido-2′,3′-dideoxythymidine (AZT; Sigma-Aldrich, St. Louis, MO) in order to inhibit reverse transcriptase (RT). After overnight incubation fresh medium with or without AZT was exchanged into the wells. Two days later, the cells were stained for avidin expression as described for viral titering and analyzed by flow cytometry.
The effect of an inhibitor of histone deacetylases on avidin expression was studied with BT4C cells 21 days posttransduction by incubating them in the presence of 5 mM sodium butyrate (Sigma-Aldrich) for 3 days. Cells were stained for avidin expression and analyzed by flow cytometry.
Quantitation of ligand-binding ability
Quantitation of the ligand-binding ability of avidin fusion protein expressed on the U-87 MG cell surface was performed with biotinylated quantum dots (bt-Qdot em605; Evident Technologies, Troy, NY) 3 days posttransduction. Cells were detached as described earlier, suspended into ice-cold binding solution containing 1% BSA, 10 mM HEPES in PBS, and incubated in the presence of 10 mM bt-Qdot for 35 min. The cells were washed and analyzed by FACS. Ligand binding was analyzed after a single dose of ligand had been delivered and after repeated delivery (after 1 and 20 hr). For redosing analysis the cells were suspended in fresh medium and incubated at 37°C for 1 hr or for 20 hr. After incubation cells were washed and bt-Qdot ligand was added as described previously. As control samples, nonbiotinylated Qdot ligands were used.
To analyze the influence of serum antibodies on ligand binding, cells were resuspended in PBS, mixed with pooled serum samples from treated animals, and incubated on ice for 30 min (20% serum). Cells were washed and bt-Qdot ligand was added as described previously.
Influence of transduction on cell viability and apoptosis
The effect of avidin fusion protein-expressing lentivirus on transduced cells was studied by measuring cell viability. HeLa, BT4C, and 293T cells were seeded on 96-well plates and transduction was performed on day 2 at multiplicities of infection (MOIs; based on p24 titer [TU/ml]) ranging from 0.1 to 1000. Cell viability was measured on day 5 with CellTiter 96 AQueous One Solution cell proliferation assay (Promega, Madison, WI). Data were expressed as percent viability compared with nontreated control cells. Similarly, caspase activity was analyzed in HeLa, BT4C, 293T, and HepG2 cells with the Apo-ONE homogeneous caspase-3/7 assay (Promega) measuring caspase-3/7 activities 2 days posttransduction at an MOI of 5 (p24-based TU/ml). Nontreated cells and cells transduced with lentivirus expressing GFP were used as controls.
Immune response in vivo
Thirty-three BDIX rats (Charles River Laboratories, Châtillon-sur-Chalaronne, France) were used in the study. Rats were distributed into five groups, of which two received injections intracranially (groups 1 and 2), two received injections intravenously (groups 3 and 4), and one served as a negative control (group 5). Repeated injections were administered 3 weeks after the initial injection to groups 2 and 4. Administration of the lentivirus (20 μl) for the intracranial group was done as described (Lehtolainen et al., 2003). For intravenous administration the rats were anesthetized with a combination of fentanyl–fluanisone (Hypnorm; VetaPharma, Leeds, UK) and midazolam (Midazolam Hameln; Hamels Pharmaceutics, Hameln, German) and vector injections were done via intravenous cannule (BD Neoflon; Becton Dickinson Infusion Therapy, Helsingborg, Sweden) into the tail vein. Because of the dead space of the syringe, 20 μl of the virus was diluted with 380 μl of saline before injection. Control serum samples were taken before virus administration from all animals. After virus administration, blood samples were collected at 1-, 3-, and 6-week time points. Blood was collected via intravenous cannule under Hypnorm- and Midazolam Hameln-induced anesthesia and Microtainer SST blood-drawing tubes (Becton Dickinson, Franklin Lakes, NJ) were used to collect blood samples. Samples were incubated at room temperature for 1 hr before centrifugation (14,000 × g, 10 min at 4°C). Serum was collected and stored at −20°C. Animals were killed by CO2 inhalation 6 weeks after virus injections.
Antibody response against avidin fusion protein
Antibody responses against avidin (IgG titers) were studied with rat sera. Combiplate 8-well Microstrips (Enhanced binding; Thermo Fisher Scientific, Waltham, MA) were coated with avidin (Sigma-Aldrich), 5 μg/ml in NaHCO3 (Honeywell Riedel-de Haën, Seelze, Germany), pH 9.5, overnight. Blocking was performed with a solution containing 1× PBS (pH 7.4), 1% BSA (Sigma-Aldrich), 500 mM NaCl (J.T. Baker, Phillipsburg, NJ), 2 mM EDTA (Sigma-Aldrich), and 0.1% Tween 20. Rat serum dilutions of 1:800, 1:3200, 1:12,800, and 1:25,600 were used. Horseradish peroxidase (HRP)-conjugated goat anti-rat secondary antibody (Abcam, Cambridge, UK) was used to detect bound serum antibodies. The substrate (0.5% 3,3′,5,5′-tetramethylbenzidine in dimethyl sulfoxide; Sigma-Aldrich) diluted 1:50 in substrate buffer (0.1 M trihydrous sodium acetate, 1.5 mM monohydrous citric acid, and 0.005% H2O2) was incubated on the wells for 15 min. The reaction was stopped with 50 μl of 2 M H2SO4 and absorbance was measured at 450 nm. Absorbances were transformed to a logarithmic scale and the end-point titers of the antibodies were calculated. The end-point titers were calculated by using a value three times the mean background as a cutoff. The averages of antibody titers at various time points were calculated as a geometric mean of end-point antibody titers.
Neutralizing assay
Serum samples from control animals and transduced animals were serially diluted in complete Dulbecco's modified Eagle's medium (Sigma-Aldrich) and an equal amount (15 μl) of serum dilution and GFP-expressing lentivirus was incubated at 37°C for 1 hr. Serum–virus dilutions were added to BT4C cells (5000 cells plated on a 96-well plate 1 day previously) and incubated overnight before changing to fresh medium. Three days after transduction the cells were trypsinized and GFP-positive cells were analyzed by FACS. Neutralization of the vector was calculated as described (Baekelandt et al., 2003).
In vitro targeting
Nanoparticles were constructed as described (Pulkkinen et al., 2008). PLA20PEG2 and biotinylated PLA20PEG2 polymers (kindly provided by J. Seppälä, Tampere University of Technology, Tampere, Finland) were diluted with paclitaxel (T-1912; Mayne Pharma, Warwickshire, UK). In vitro targeting was performed on 96-well plates. Each well was coated with a total of 10,000 BT4C glioma control cells or cells expressing avidin fusion protein (sorted cells; ∼60% positive). Twenty-four hours later the cells were exposed for 30 min to nanoparticles carrying paclitaxel. Cells were then washed twice with PBS. Cell viability was measured 72 hr after the treatment by CellTiter-Blue assay (Promega). Transduced BT4C cells treated with nonbiotinylated nanoparticles and nontransduced BT4C cells treated with biotinylated nanoparticles, nonbiotinylated nanoparticles, or paclitaxel alone were used as controls. Paclitaxel concentrations used in the experiment were 50 and 100 μg/ml.
Avidin fusion protein expression and cellular immune response in vivo in rat malignant glioma model
BDIX rats (Charles River Laboratories) were anesthetized with a solution of ketamine and medetomidine hydrochloride administered intraperitoneally. A total of 10,000 BT4C cells in a volume of 10 μl was injected into the right corpus callosum as described (Tyynela et al., 2002). Lentivirus (2 × 15 μl; total of 2.5 × 107 p24-based TU) was injected 3 weeks later, using the same coordinates. Five days after the transduction animals were killed and perfused with PBS. Brains were removed and fixed with 4% paraformaldehyde (PFA) for 6 hr. Paraffin sections were immunostained with goat anti-avidin (diluted 1:100). The cellular immune response in the brain sections was studied by staining with CD68 (mouse anti-rat CD68, also known as ED1; AbD Serotec, Oxford, UK) and CD8 (mouse anti-rat CD8α; AbD Serotec) antibodies, using 1:500 dilutions. Secondary antibody horse anti-mouse IgG (Vector Laboratories), a VECTASTAIN ABC kit (Vector Laboratories), and 3,3′-diaminobenzidine (DAB; Invitrogen Zymed, South San Francisco, CA) were used. Counterstaining was performed with Harris hematoxylin. Sections were imaged by Olympus BX-41 microscopy.
Statistical analyses
Statistical analyses were performed with GraphPad Prism version 5 (GraphPad Software, San Diego, CA). Data were analyzed by one-way analysis of variance (ANOVA) and Kruskal–Wallis test, with Dunn's multiple post hoc test used in the comparisons; or by two-way ANOVA and the Bonferroni post hoc test. The α level for significance was set p < 0.05.
Results
Characterization of avidin fusion protein-expressing lentivirus
A gene encoding Lodavin, a fusion protein consisting of avidin and the endocytotic part of the LDLR (Lehtolainen et al., 2003), was cloned into a lentiviral plasmid and recombinant lentiviruses were produced. Four different titration methods (p24 ELISA, viral RNA RT-qPCR, FACS, and proviral DNA qPCR) were applied to assess vector production (Table 1). The p24 titers of batches ranged from 8.1 × 1011 to 2.5 × 1012 viral particles (VP) per milliliter whereas the conversion of these values to transducing units per milliliter yielded 1.6–4.1 × 109 TU/ml. The RNA titers of the produced batches were in the range of 6.5 × 108 to 1.1 × 109 VP/ml. The functional titers measured by FACS ranged from 1.0 × 107 to 1.5 × 107 TU/ml in U-87 MG cells and from 1.7 × 107 to 5.2 × 107 TU/ml. Overall, the titers in rat glioma cells were 2- to 4-fold higher compared with human glioma cells. DNA titer measured as the number of integrated proviral copies yielded a titer of 1.2 × 108 for viral batch IV, which was analyzed. Altogether the results showed that avidin fusion protein-expressing lentivirus had slightly decreased titers compared with an average control virus with functional titers ranging from 108 to 109. On the basis of our results, the estimated ratio of total to functional particles was 1:60–1:200,000, indicating the presence of defective viral particles. No RCL was detected in any of the analyzed lentivirus preparations by the p24 ELISA-based method.
Abbreviations: FACS, fluorescence-activated cell sorting; NA, not analyzed; qPCR, quantitative PCR; TU, transducing units; VP, viral particles.
Results are given from two independent experiments.
Expression of avidin fusion protein
The total (tE) and surface (S) expression of avidin fusion protein was quantified from transduced U-87 MG cells by anti-avidin staining. The percentage of cells expressing the fusion protein in the cytoplasm and on the cell membrane (tE%; average, 50.3%) was compared with the percentage of cells expressing the fusion protein on the cell surface only (S%; 24.5%) (Fig. 1A). Cell surface expression represented about 50% of total expression, S/tE (%). However, comparison of the mean fluorescence indexes (tE average, 3550; S average, 500), indicating the amount of avidin fusion protein expressed, showed that 14% of the expressed protein was found on the glioma cell surface (Fig. 1B). In addition, transduced cells were plated on coverslips and visualized by fluorescence microscopy after immunostaining (Fig. 1C–F). The difference between total expression (Fig. 1C) and surface expression (Fig. 1E) was clearly visible by microscopy: total expression was bright whereas surface expression was seen with less intensity.

Total and surface expression of avidin fusion protein in glioma cells transduced with recombinant lentivirus. (
The duration of avidin fusion protein expression was monitored for more than 30 days in rat glioma cells (Fig. 1G and H). A decline in the percentage of avidin-expressing cells from 90–100 to 20–30% was seen with the two virus batches studied, after which expression stabilized. Similar results were obtained with U-87 MG human glioma cells. However, when avidin fusion protein-expressing cells were separated from negative cells (sorted) 3 days posttransduction, the percentage of positive cells settled at 55% (Fig. 1G). Also, a decrease in protein expression levels was seen as indicated by the drop in mean fluorescence intensities (Fig. 1H) and Western blot (data not shown). The FACS and DNA titers on day 10 were 5- and 45-fold lower, respectively, compared with day 3 levels (Fig. 1I). Incubation of the cells in the presence of RT inhibitor AZT revealed that 14–28% of avidin expression on day 3 came from pseudotransduction (Fig. 1J). Treatment with sodium butyrate (5 mM), an inhibitor of histone deacetylases, had no effect on avidin fusion protein expression on day 24 posttransduction, indicating that no epigenetic gene silencing occurs in vitro (data not shown).
Biotin-binding capability of avidin fusion protein after readministration
The biotin-binding capacity of avidin fusion protein on U-87 MG human glioma cells was studied with biotinylated quantum dots (bt-Qdot). Binding of bt-Qdot was measured at time point 0 and readministration was performed 1 and 20 hr later (Fig. 2). Readministration of the ligand after 1 hr showed a residual ligand-binding capacity of 20–50% whereas 65–85% of the initial binding capacity was seen at 20 hr. No residual signal from the first administration was detected at 1–20 hr without the second dose of bt-Qdot, indicating that internalization of the Qdots at 37°C led to the loss of fluorescence signal (data not shown). This is supported by previous studies, which have shown that Lodavin is indeed endocytosed and recirculated (Lehtolainen et al., 2003).

Biotin-binding capacity of avidin fusion protein after repeated administration of biotinylated ligands. The percentage of positive cells binding the bt-Qdot ligand at first administration (time point 0), and after delivery of the second ligand at 1 and 20 hr, was measured by FACS. I, II, and II represent individual batches of lentiviral vector. Results are presented as averages ± standard deviation of triplicates.
Effect of transduction on target cells
The influence of avidin fusion protein on transduced HeLa, BT4C, and 293T cells was studied by measuring cell viability (Fig 3). No effect of lentivirus transduction on cell growth was seen at lower MOIs, using cell proliferation as the determinant. However, when the MOI was more than 50, a decrease in cell viability was detected. The most probable reason for this is the toxicity of the vesicular stomatitis virus G protein (VSV-G) on the viral envelope, which has been noticed after virus transduction at high viral concentrations (Park et al., 2000; Watson et al., 2002). In addition, GFP showed some decrease in cell viability compared with the avidin fusion protein in BT4C cells at high MOIs. This may be due to the toxicity of GFP (Liu et al., 1999). Thus far avidin has not been shown to have any toxicity when injected into patients (Paganelli et al., 1991). These studies proved that avidin fusion protein does not have any considerable effect on the viability of transduced cells. In line with this, caspase-3/7 activity (apoptosis) could not be detected in the transduced cells (data not shown).
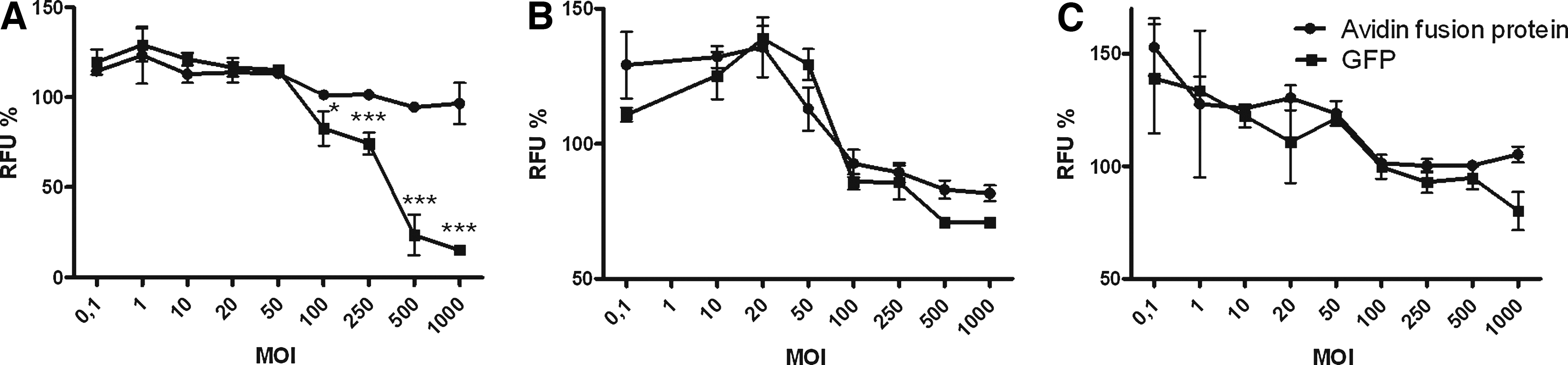
Toxicity of lentivirus transduction. Viability of (
Antibody response against avidin fusion protein
The immune response against the avidin fusion protein was evaluated by avidin ELISA. Lentivirus was administered intracranially or intravenously and 3 weeks later the administration was repeated with half the animals. Serum samples were collected 3 and 6 weeks after the first administration and the formation of avidin antibodies was analyzed in all serum samples (Fig. 4A and B). An increase in the average titers was detected at the 6-week time point compared with the samples taken at 3 weeks. The antibody titers were also found to be higher in the intracranial group compared with the intravenous group at the 3-week time point However, the differences were not statistically significant in any of the groups (Kruskal–Wallis, p = 0.0714). This showed that antibodies are produced against the transgene product and that repeated administration did not significantly boost antibody production.

Antibodies against the transgene product were determined from the serum samples 3 and 6 weeks after virus administration (
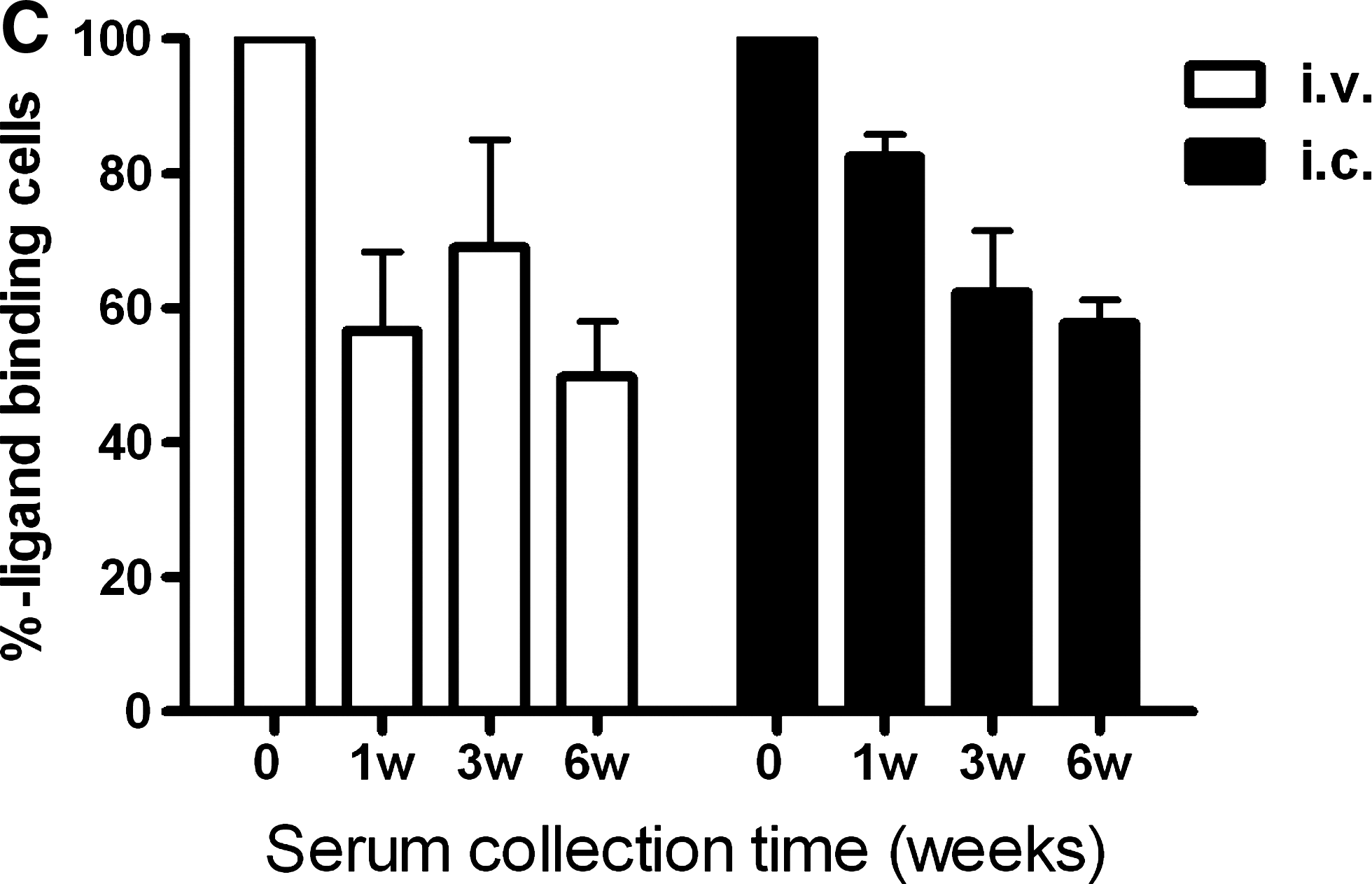
The influence of antibodies on biotin-binding capacity was also studied (Fig. 4C). Serum samples taken 1, 3, and 6 weeks after virus administration (done intravenously or intracranially) were incubated with the transduced cells before ligand administration. Biotin-binding capacity was decreased by 50%.
Neutralizing antibodies against viral vector
To investigate whether treated animals developed neutralizing antibodies against the lentiviral vector, a neutralization assay was performed in vitro. Incubation of LV-GFP vector with serum dilutions (1:100 and 1:500) from virus-injected animals (3- and 6-week time points) inactivated the virus to such an extent that no transduction occurred (Fig. 5). With a 1:3000 serum dilution neutralization was not complete. No difference in transduction efficiency was detected between the control virus incubated in the presence of serum samples collected before vector administration or no serum. No significant difference was found between the single and repeated administration groups.

Serum neutralization assay. GFP-expressing lentivirus was incubated in the presence of serum samples (dilutions 1:100, 1:500, and 1:3000) collected 3 and 6 weeks after virus administration (
In vitro targeting, using avidin fusion protein
To determine whether avidin fusion protein can be used for targeted cancer therapy, biotinylated nanoparticles carrying paclitaxel (Pulkkinen et al., 2008) were targeted to transduced glioma cells in vitro. Cells were treated with nanoparticles carrying increasing concentrations of paclitaxel and cell viability was measured 72 hr later by CellTiter-Blue assay. After the treatment only 9.9 or 8.2% of lentivirus-transduced cells were alive when 50- or 100-μg/ml concentrations of biotinylated nanoparticles carrying paclitaxel were used, respectively (Fig. 6). For comparison, 32.0 or 13.0% of lentivirus-transduced cells were alive when incubated with nonbiotinylated nanoparticles carrying the same concentration of paclitaxel. Paclitaxel alone resulted in 30–40% cell viability.
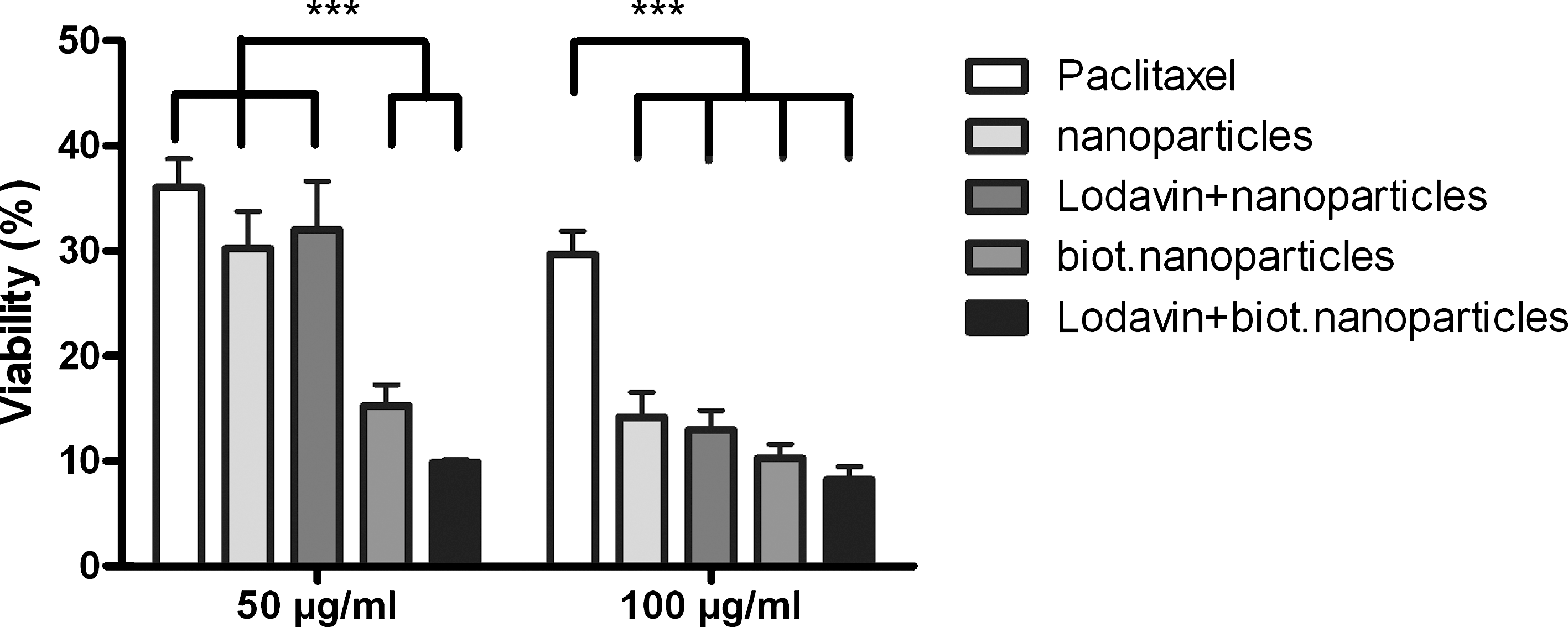
Viability measurements of BT4C control cells and cells expressing avidin fusion protein (∼60% positive cells) after a 30-min exposure to paclitaxel-carrying nanoparticles at either (left) 50 or (right) 100 μg/ml. Cell viability was measured 72 hr after treatment. Data were analyzed by two-way ANOVA with Bonferroni post hoc test. *** p < 0.001. For each group, n ≥ 4 (average).
In vivo expression of avidin fusion protein and cellular immune response
The expression of avidin fusion protein was also studied in vivo in a rat glioma model (Fig. 7A–C). Lentivirus was injected into tumors and 5 days later animals were killed and brain tissue samples were collected and stained with anti-avidin antibodies. Expression was detected in the area of the tumor (Fig. 7A and B). These results confirmed the feasibility of lentivirus-mediated gene delivery to brain tumors by local administration. The cellular immune response was also studied by immunostaining the brain sections with CD8 and CD68 antibodies. Accumulation of macrophages, microglial cells, and CD8+ T cells was seen in tumor, which might implicate the immune response caused by transgene or vector preparation (Fig. 7D and E). However, accumulation in nontransduced tumor was also seen (Fig. 7F and G), which is in line with the previous studies showing that tumors are extensively infiltrated by the immune response (King et al., 2008).

Expression of avidin fusion protein in rat glioma tumor model, detected by anti-avidin staining. (
Discussion
The avidin–endocytotic LDLR fusion protein, Lodavin, is designed as a transmembrane protein that mediates the binding of biotinylated pharmaceutical conjugates to the cell membrane (Lehtolainen et al., 2003). This two-step (gene transfer followed by a biotinylated drug) pretargeting strategy based on avidin–biotin technology enables the concentration of drugs in specific cells or tissues while minimizing side effects to nontarget organs and eliminating the need to engineer specific antibodies for each application (Goodwin and Meares, 2001). In this study, Lodavin-expressing lentivirus was constructed and the efficiency of lentivirus production was confirmed by four titration methods. The RNA and p24 ELISA methods yielded significantly higher titers and were thus found to be less reliable in predicting the functional titer (Geraerts et al., 2006). The p24 ELISA, in particular, grossly overestimated the titers, probably because of the variable amount of free non-particle-associated p24 proteins (Geraerts et al., 2006). Of the functional titers, the DNA titer was 12-fold higher than the FACS titer, which could be due to pseudotransduction, poor detection of avidin expression by FACS, or, as previously suggested, to the fact that not all proviral DNA molecules correspond to transcriptionally active forms (Sastry et al., 2002).
To demonstrate the potential of our targeting approach for targeted delivery of biotinylated ligands we analyzed the expression levels of the avidin fusion protein in detail and studied its ligand-binding capability on human glioma cells. Moreover, the feasibility and time scale of repeated ligand delivery were analyzed. Clarification of these parameters enables selection of optimal time frame(s) of radiopharmaceutical ligand (and/or ligand-enabling imaging pretherapy) delivery and assessment of estimated therapy outcome. The results showed that 50% of the cells expressing avidin fusion protein displayed it on the cell surface and that surface expression represented 14% of the total expression level at a given moment. These results can be explained by the endocytic cycling of the avidin–LDLR fusion protein (Lehtolainen et al., 2003), which is like that of the LDLR, in that part of the receptors are internalized at any given instant (Hao and Maxfield, 2000). Interestingly, the results demonstrated that avidin expression on day 3 partly resulted from pseudotransduction due to plasmid or protein transfer. Stable gene expression corresponding to expression from integrated proviral DNA was attained 10–20 days posttransduction. As transgene expression resulting from pseudotransduction has been found to disappear as soon as day 4 posttransduction results suggests that transient expression from the unintegrated viruses also contributes to some level of avidin fusion protein expression at earlier time points (Haas et al., 2000; Nash and Lever, 2004). This was supported by a drop in DNA titer from day 3 to day 10 (45-fold). To further support this, the functional FACS and DNA titers yielded similar results on day 10, whereas on day 3 the difference was still 12-fold. Optimization of virus production and purification is needed to reduce the extent of pseudotransduction and to minimize the amount of integration-defective viral particles.
The ligand-binding studies indicated that a second ligand administration is feasible ≥1 hr after the first administration, but that a better binding response can be obtained ≥20 hr. This could be expected as the LDLR recycling time is fast, with a half-time of 9 to 12 min (Hao and Maxfield, 2000). These results suggest that a second ligand administration can be performed within a day of the first treatment. Moreover, as long-term expression of avidin fusion protein was achieved, multiple administrations over a broad range of time can be envisioned to improve the overall efficiency of the pretargeting therapy.
One of the most important factors regarding the safety of gene medicines is the immunogenicity of the vector (Raty et al., 2008). Immune responses can lead to the elimination of both the vector and the transduced cells, decreasing the effect and duration of treatment (Bessis et al., 2004). Another factor is the immune response against the transgene. In general, lentiviruses are considered to be mildly immunogenic (Bessis et al., 2004; Follenzi et al., 2007) but may still give rise to immune responses. These responses may be more related to insufficient purification during the manufacturing of vector than to the components of the vector (Baekelandt et al., 2002, 2003; Follenzi et al., 2007). However, VSV-G has been shown to be immunogenic and lentiviral particles are inactivated by human serum complement after systemic delivery (DePolo et al., 2000; Schauber-Plewa et al., 2005). Antibody responses to the virus itself and transgene product (avidin) were studied here. As avidin targeting can be used for other applications than tumor targeting both the intracranial and intravenous administration routes were studied. Readministration of the vector was performed to determine the influence and strength of immune responses after rechallenge. All the treated animals developed neutralizing antibodies against the vector, in line with previous publications (Baekelandt et al., 2003). The antibodies produced probably inhibited transduction during the second administration. Therefore, the repetition of immunization with the same vector is not feasible and to overcome this another pseudotype should be used for the second administration (Cronin et al., 2005). The virus used was concentrated by ultracentrifugation but, in addition, a more effective downstream process (Rodrigues et al., 2007) would be essential to remove impurities from the vector preparation and thus to minimize the formation of neutralizing antibodies in future (Baekelandt et al., 2003). In addition, less immunogenic pseudotypes (Cronin et al., 2005) or the incorporation of complement-regulatory proteins into the vector particles (Schauber-Plewa et al., 2005) might be considered to prevent the generation of an immune response. On the other hand, in cancer therapy the immune response at the area of tumor might be advantageous in eliminating cancer cells from the body.
The production of antibodies against the transgene (avidin) was also demonstrated. This is in line with previous studies showing immunoreactivity of avidin with cancer patient serum samples exposed to avidin (Hytonen et al., 2003). The results indicated that the average antibody titers in the intracranial group were somewhat higher compared with the average for the intravenous group. This could be explained by the faster clearance of the virus by the complement system after intravenous delivery (DePolo et al., 2000). No boost in antibody response was detected after virus readministration, which can be explained by neutralizing antibodies preventing virus transduction and thus avidin expression.
Avidin antibodies can affect the repeated use of pretargeting therapies (Hytonen et al., 2003). Therefore, we wanted to study the influence of avidin antibodies on ligand-binding efficacy by incubating transduced cells with immunized serum before ligand administration. The results showed that the antibodies decreased ligand binding by 50%, thus proving that the antibody response decreases but does not completely inhibit multiple ligand administrations.
The potential use of avidin fusion protein, delivered via lentiviral vector, for cancer targeting was confirmed in vitro with biotinylated nanoparticles carrying paclitaxel. In a previous study, biotinylated paclitaxel was successfully used in three-step targeting using avidin–biotin technology (Pulkkinen et al., 2008). In our two-step approach, biotinylated nanoparticles were more potent in killing transduced cells than nontargeted nanoparticles or paclitaxel alone. This difference was, however, significant only at the lower paclitaxel concentration (50 μg/ml). This could be due to the significant cytotoxicity caused by the nanoparticles filled with higher concentrations (100 μg/ml) of paclitaxel. Cell intake of biotinylated nanoparticles alone was also significant, which might be due to binding of biotin to the multivitamin transporters on the cell surface (Zempleni, 2005). Finally, lentivirus-mediated gene delivery to rat glioma cells was confirmed in vivo, thus providing the groundwork for more extensive animal studies examining survival benefit. Even though tumor targeting was used here to provide the proof-of-principle of the concept, other applications for the treatment of cardiovascular or inflammatory diseases can also be envisaged.
In conclusion, we have created a novel lentiviral vector for long-term expression of avidin fusion protein on the surface of cells destined for two-step pretargeting therapies. Expression of avidin fusion protein on the cell surface was demonstrated and subsequent binding of biotinylated ligands was achieved. Moreover, repeated administration of the ligand was proven to be feasible. This lentivirus-mediated delivery of the avidin fusion protein could offer significant improvements for targeted therapy of cancer and other diseases.
Footnotes
Acknowledgments
The authors thank Siiri Väistö, Juha Ruuskanen, Anne Martikainen, Joonas Malinen, and Riikka Eisto for excellent technical assistance, and are grateful to Antti Ropponen (Department of Clinical Microbiology, Kuopio University) for the performance of cell sorting. This study was supported by Ark Therapeutics Group and the Finnish Academy.
Author Disclosure Statement
The authors Hanna Lesch, Jere Pikkarainen, Minna Kaikkonen, Miia Taavitsainen, Haritha Samaranayke, Pauliina Lehtolainen, Taina Vuorio, and Ann-Marie Määttä are employees of Ark Therapeutics. No competing financial interests exist for Thomas Wirth, Kari Airenne, and Seppo Ylä-Herttuala.