
Abstract
To improve the biocompatibility of a gene vector and to avoid its being eliminated by the immune system, polyethylenimine (PEI) was modified with poly(ethylene glycol) (PEG) before G250 monoclonal antibody (mAb) conjugation. G250-PEI-PEG was capable of forming complexes with DNA in the right size distribution, and the G250 mAb modification significantly improved PEI transfection of G250-positive cells. The highest transfection efficiency was seen in HeLa cells as determined by flow cytometry after transfection with the gene encoding green fluorescent protein: 2-fold higher compared with the transfection of HepG2 cells. Blocking the surface antigen on the cell membrane of HeLa cells by incubation with free G250 mAb, or by downregulating G250 expression by small interfering RNA transfection, resulted in a remarkable decrease in transfection efficiency. These data indicate the targeting effect of G250 antibody modification. The presence of serum decreased transfection efficiency in a concentration-dependent manner. However, the transfection of HeLa cells with G250-PEI-PEG remained significant in the presence of 30% serum. In an in vivo study, G250-PEI-PEG exhibited high transfection efficiency in tumors. In addition, pathological analysis did not show obvious toxicity caused by the materials used. These suggest that PEG- and G250 mAb-modified PEI could be a useful nonviral gene vector for in vivo study.
Introduction
To overcome these problems, some research groups have synthesized various poly(ethylene glycol)-grafted PEIs (PEG-PEIs) and studied the biophysical and biological properties of the polymer/DNA complexes (Erbacher et al., 1999; Nguyen et al., 2000; Kunath et al., 2002; Petersen et al., 2002a). Because of the high solubility of PEG in water, its nonimmunogenicity, and good biocompatibility, it has been used to modify PEI to improve its application in many gene delivery systems (Sung et al., 2003).
Modification with antibodies has been a strategy for targeted gene delivery. Incorporation of antibodies into PEI made it possible to target certain tissues (Torchilin et al., 1992; Blume et al., 1993) and tumors (Maruyama et al., 1997). G250, as one of the most extensively studied monoclonal antibodies associated with the HeLa cell line, has been well studied (Uemura et al., 1999). G250 incorporated into PEI will be able to specifically target G250 antigen-positive tumor cells (e.g., HeLa cells) while minimizing gene delivery to normal tissues because no G250 expression has been detected in normal tissues.
In this study, PEI is modified with PEG before G250 conjugation in order to improve the biocompatibility of PEI, and to avoid its being eliminated by the immune system. We hypothesize that PEG and G250 modification will improve the biocompatibility of PEI, target tumor cells that overexpress G250 antigen, and enhance transfection efficiency in the presence of serum and in vivo.
Materials and Methods
Materials
Polyethylenimine (PEI, branched; molecular mass, 25 kDa), PEG monomethyl ether (PEG) 550 Da, PEG 2 kDa, hexamethylene diisocyanate (HMDI) (≥99%), ethanol, 2-iminothiolane hydrochloride, Sephadex G-75, and 3-(2-pyridyldithio)propionic acid N-hydroxysuccinimide ester [synonym, N-succinimidyl 3-(2-pyridyldithio)propionate, SPDP] were purchased from Sigma-Aldrich (St. Louis, MO). Chloroform (Sigma-Aldrich) was treated with HMDI for 4 hr at 60°C and distilled to remove any traces of water. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Invitrogen (GIBCO, Grand Island, NY). The G250 monoclonal antibody (G250) was prepared in our laboratory as previously described (Duan et al., 2008). HeLa and HepG2 cell lines were kindly provided by Huaxi Medical College (Sichuan University, Chengdu, China). The NIH/3T3 cell line was obtained from the Chinese Type Culture Collection (CTCC, Wuhan University, Wuhan, China). The plasmid pEGFP-C1, kindly provided by C. Song (Institute of Biomedical Engineering, Chinese Academy of Medical Sciences, Tianjin, China), was prepared in Escherichia coli DH5α cells and isolated with a QIAfilter plasmid giga kit (Qiagen, Hilden, Germany). The plasmid pGL3, Glo lysis buffer, and the Bright-Glo luciferase assay system were purchased from Promega (Madison, WI). Carbonic anhydrase IX (CA IX) siRNA was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All cell culture media and supplies were purchased from GIBCO. BALB/c-nu/nu athymic mice (female, 6 weeks old) were purchased from the Laboratory Animal Center of the Academy of Military Medical Sciences (Beijing, China). The animal studies were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals (Tianjin, China; revised in June 2004) and adhered to the Guiding Principles in the Care and Use of Animals published by the American Physiological Society (Bethesda, MD).
Cell culture
HeLa, HepG2, and NIH/3T3 cells were used in this study. HeLa cells express a high level of G250 antigen, whereas HepG2 cells express a minimal level. HeLa, HepG2, and NIH/3T3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM). Medium was supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml).
Preparation of PEI-PEG copolymers
The preparation of PEI-PEG copolymers was carried out according to the previous report and in accordance with its synthesis scheme (Petersen et al., 2002a). PEG 550 Da and 2 kDa, dissolved in chloroform at a concentration of 200 g/liter, were activated for reaction with the amino groups of PEI with a 10-fold excess of HMDI at 60°C for 24 and 48 hr, respectively. HMDI excess was carefully removed by extraction three times with light petrol. Activated PEGs were reacted with PEI at concentrations of 10 g/liter in chloroform at 60°C for 24 and 48 hr, respectively. The reaction solutions were concentrated to 100 g/liter by evaporation of the solvent and dropped into a 20-fold larger volume of diethyl ether to obtain the copolymer by precipitation. Last, the products were dried in vacuum. Nuclear magnetic resonance (NMR) spectroscopy was used to characterize the polymers on the basis of their 1H spectrum.
Preparation of G250-PEI-PEG conjugates
SPDP (10 mM in 20 μl of dimethyl sulfoxide [DMSO]) was added into the PEI-PEG solution (25 mg in 5 ml of phosphate-buffered saline [PBS]). The reaction was carried out at room temperature for 24 hr on a shaker, and terminated by adding 100 μl of glycine (1 mol/liter). G250 antibody (0.375 mg), preactivated by the addition of 21.8 μl of 2-iminothiolane hydrochloride (20 μM), was added into the reaction (Thorpe et al., 1987), and the reaction was continued for 24 hr. The product was first separated by passage through a Sephadex G-75 column (fractionation range, 3000–70,000), dialyzed against deionized water, and then lyophilized.
Gel retardation assay
A gel retardation assay was performed to examine the ability of G250-PEI-PEG to condense plasmid pEGFP-C1. DNA was mixed with G250-PEI-PEG in PBS at weight ratios varying from 0.5 to 3.0. The mixtures were incubated at room temperature for 30 min and then subjected to electrophoresis on 0.8% agarose gels containing ethidium bromide (0.4 μg/ml). Images were recorded with an ImageMaster VDS (Pharmacia, Uppsala, Sweden).
Laser light-scattering assay
G250-PEI-PEG/DNA complexes were prepared at room temperature for 30 min. The optimal weight ratios for G250-PEI-PEG were chosen on the basis of the results of the gel retardation assays. Samples of the complexes were filtered (pore size, 0.45 μm) and analyzed with a light-scattering spectrometer (BI-200SM; Brookhaven Instruments, Holtsville, NY), equipped with a digital correlator (BI-9000AT; Brookhaven Instruments), at 532 nm to determine the granulometric distribution of the DNA complexes.
Cytotoxicity assay
Cytotoxicity of the unconjugated PEI, PEI-PEG, and G250-PEI-PEG was evaluated by MTT assay. NIH/3T3 cells were seeded onto 96-well plates at a density of 104 cells per well and incubated for 24 hr. The filtered (pore size, 0.22 μm) samples were then added into the cells. After incubation for another 24 hr, 25 μl of MTT solution (5 mg/ml in PBS) was added to each well. Four hours later, the medium containing MTT was removed and the samples in the wells were air dried. Acidic isopropanol (100 μl, 0.04 M HCl in absolute isopropanol) was added to dissolve the formazan crystals. The optical density of the solution was measured at 570 nm, using a microplate reader (Multiskan Ascent; Labsystems, Helsinki, Finland).
In vitro transfection
Cells were seeded at a density of 105 cells per well in 24-well plates and incubated for 24 hr. The medium was replaced with serum-free medium one-half hour before transfection. Two micrograms of DNA and PEI, PEI-PEG, or G250-PEI-PEG was diluted in 100 μl of serum-free medium separately. DNA solution was added into PEI or IgG-PEI solution, mixed well, and incubated at room temperature for 30 min. The complexes were then added into the wells (2 μg of DNA per well). Four hours later, the medium was discarded and replaced with complete medium. At 48 hr, green fluorescent protein (GFP)-positive cells were viewed under an inverted fluorescence microscope and images were recorded with a charge-coupled device (CCD) (ECLIPSE TE2000-U; Nikon, Tokyo, Japan). For the flow cytometry assay (Altra FCM; Beckman Coulter, Fullerton, CA), cells were trypsinized and analyzed. The transfection efficiency was expressed as the percentage of GFP-positive cells. Efficiency was analyzed by fluorescence-activated cell sorting (FACS). To identify the targeting effect of G250 monoclonal antibody (mAb) modification, HeLa cells were treated with either G250 mAb (Duan et al., 2008) or siRNA before transfection. In the siRNA treatment, siRNA was delivered to HeLa cells by means of the transfection reagent provided with the kit from Santa Cruz Biotechnology. Twenty-four hours later transfection with G250-PEI-PEG(550)10 (the index stands for the average number of PEG blocks bound to one PEI molecule) was performed. To test the effect of serum on transfection, transfection was performed in the presence of 20 and 30% serum.
In vivo transfection
Six-week-old BALB/c-nu/nu athymic mice (female) were kept in groups of eight and had free access to food and water. Mice were injected subcutaneously via the flank with 5 × 107 HeLa cells and, approximately 5 weeks later, tumor nodules 10 to 15 mm in maximal diameter were obtained in about 90% of the animals (Miyatake et al., 1999). The animals were randomly divided into four groups: PBS (control), PEI, PEI-PEG, and G250-PEI-PEG. The polymer/DNA complexes were prepared and incubated for 30 min before injection. For each animal, 200 μl of DNA complexes containing 40 μg of DNA was injected into the tumor. Transfection and expression were analyzed by both inverted fluorescence microscopy and luciferase activity assay.
Six days after transfection, tumors were harvested and embedded in O.C.T. medium (Sakura Finetek USA, Torrance, CA). Frozen sections (7 μm) were cut and viewed for GFP fluorescence. At the same time, for the luciferase assay, tumor tissue was frozen in liquid nitrogen and then the frozen tissue was ground with a mortar and pestle chilled with liquid nitrogen. Lysis buffer (500 μl) from the luciferase assay kit was added, and then the solution was subjected to three freeze–thaws (between liquid nitrogen and a 37°C water bath). The lysate was centrifuged in a microcentrifuge (10,000 × g for 3 min) and the supernatant was kept. Lysis buffer (500 μl) was added to the pellet, after which the freeze–thaws were repeated and the lysate was centrifuged in a microcentrifuge (10,000 × g for 3 min). The supernatant was pooled with the supernatant recovered previously (for a total of 1 ml of lysate) (Manthorpe et al., 1993). Fifty microliters of the lysate was mixed with an equal volume of luciferase substrate; the luminescence was measured immediately with a Mini-Lum luminometer (Bioscan, Washington, D.C.). Relative light units were standardized for protein concentration, which was determined by bicinchoninic acid (BCA) protein assay (Nanjing KeyGen Biotech, Nanjing, China).
Statistical analysis
A paired-samples t test was used in this study. p < 0.05 was considered significant.
Results
Preparation of PEI-PEG copolymers and G250-PEI-PEG conjugates
PEI-PEG(550)5, PEI-PEG(550)10, and PEI-PEG(550)20, as well as PEI-PEG(2k)3, PEI-PEG(2k)5, PEI-PEG(2k)10, and PEI-PEG(2k)15, were prepared at various weight ratios and their transfection efficiencies were evaluated in HeLa cells (Figs. 1 and 2). There was no significant difference in efficiency between PEI-PEG(550)5 and PEI-PEG(550)10. In addition, there was almost no difference in efficiency between PEI-PEG(2k)5 and PEI-PEG(2k)10. PEI-PEG(550)10 and PEI-PEG(2k)10 were chosen to prepare the G250-PEI-PEG conjugates because of their lower cytotoxicity (data not shown). The weight ratios (polymer to DNA) are important for transfection efficiency. As shown in Figs. 1 and 2, the optimal weight ratios for PEI-PEG(550) and PEI-PEG(2k) were 5:1 and 50:1, respectively. This difference was probably due to the modification of PEI with bigger PEG molecules, which diluted the charge density of PEI, and much higher weight ratios were therefore required for sufficient complexation with DNA.
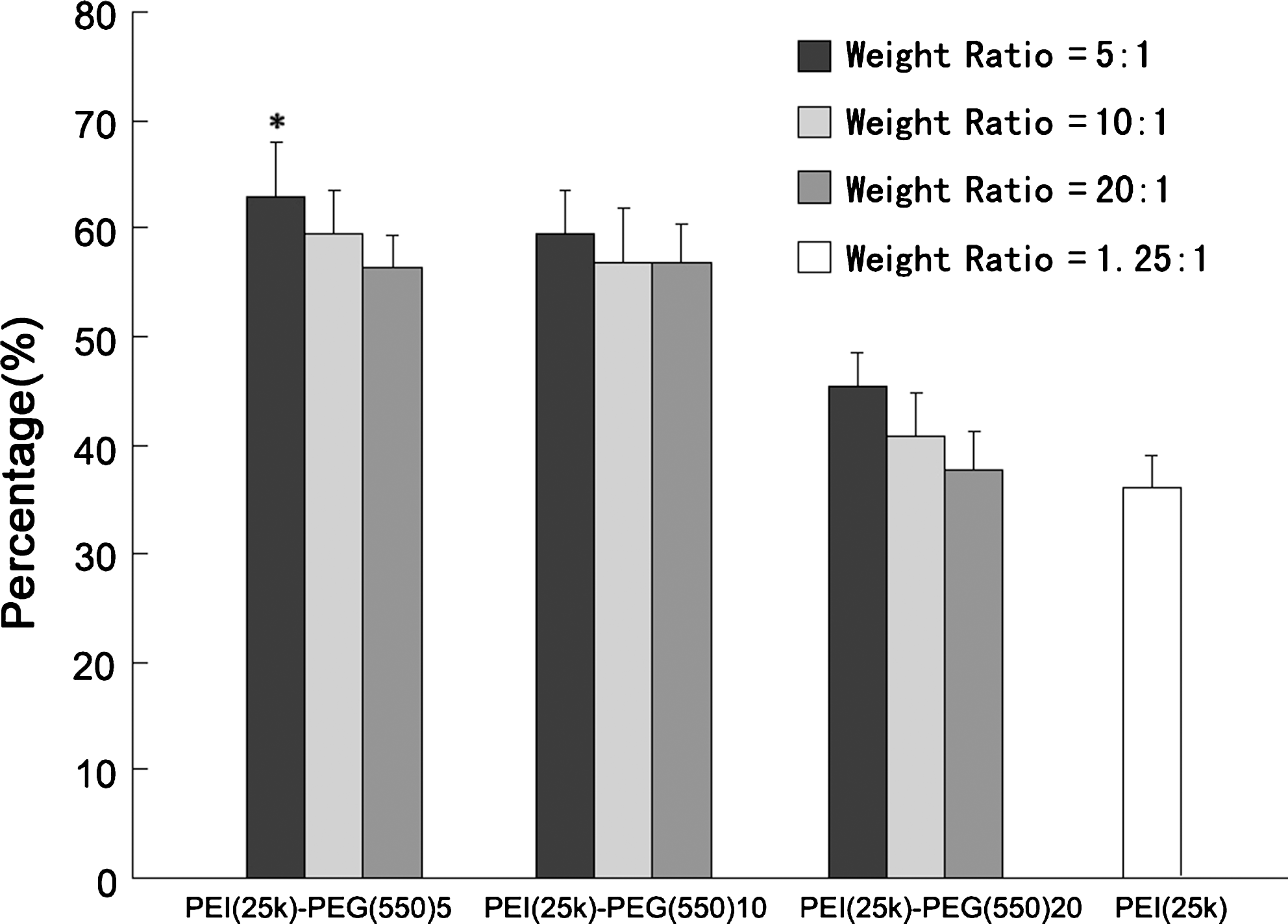
Transfection efficiency of PEI-PEG(550), analyzed by fluorescence-activated cell sorting (FACS). The transfection of HeLa cells by PEI-PEG(550)5 and PEI-PEG(550)10 at a weight ratio of 5:1 is higher than at other ratios, and it is significantly higher than with PEI (*p < 0.05).

Transfection efficiency of PEI-PEG(2k), analyzed by FACS. The transfection of HeLa cells by PEI-PEG(2k)5 and PEI-PEG(2k)10 at a weight ratio of 50:1 is slightly higher than at other ratios. All the PEI-PEG(2k) conjugates have lower transfection compared with PEI.
Gel retardation assay
The formation of G250-PEI-PEG/DNA polyplexes as a function of weight ratio was determined by gel retardation assay. The weight ratio of G250-PEI-PEG/DNA polyplexes reflects the overall positive-to-negative charge balance of the complexes. Figure 3A and B shows that G250-PEI-PEG(550)10 and G250-PEI-PEG(2k)10 could completely eliminate the mobility of plasmid DNA at weight ratios of 1 and 2, suggesting that they are capable of forming polyplexes.

Characterization of DNA polyplexes. Shown is the electrophoresis of (
Laser light-scattering assay
The granulometric distribution of the complexes was tested with the laser light-scattering detector after incubation of G250-PEI-PEG(550)10 or G250-PEI-PEG(2k)10 with plasmid DNA for 30 min at room temperature. As shown in Fig. 3C and D, the scope of G250-PEI-PEG(550)10/DNA ranged from 83.5 to 157.5 nm. The scope of G250-PEI-PEG(2k)10/DNA ranged from 143 to 217 nm.
Cytotoxicity assay
The cytotoxicity of PEI-PEG copolymers, G250-PEI-PEG conjugates, and unconjugated PEI was tested in NIH/3T3 cells by MTT assay. The viability of the cells decreased with an increase in polymer concentration. However, the viability of G250-PEI-PEG-treated cells was much greater at all polymer concentrations compared with cells treated with PEI or PEI-PEG copolymers (Fig. 4).
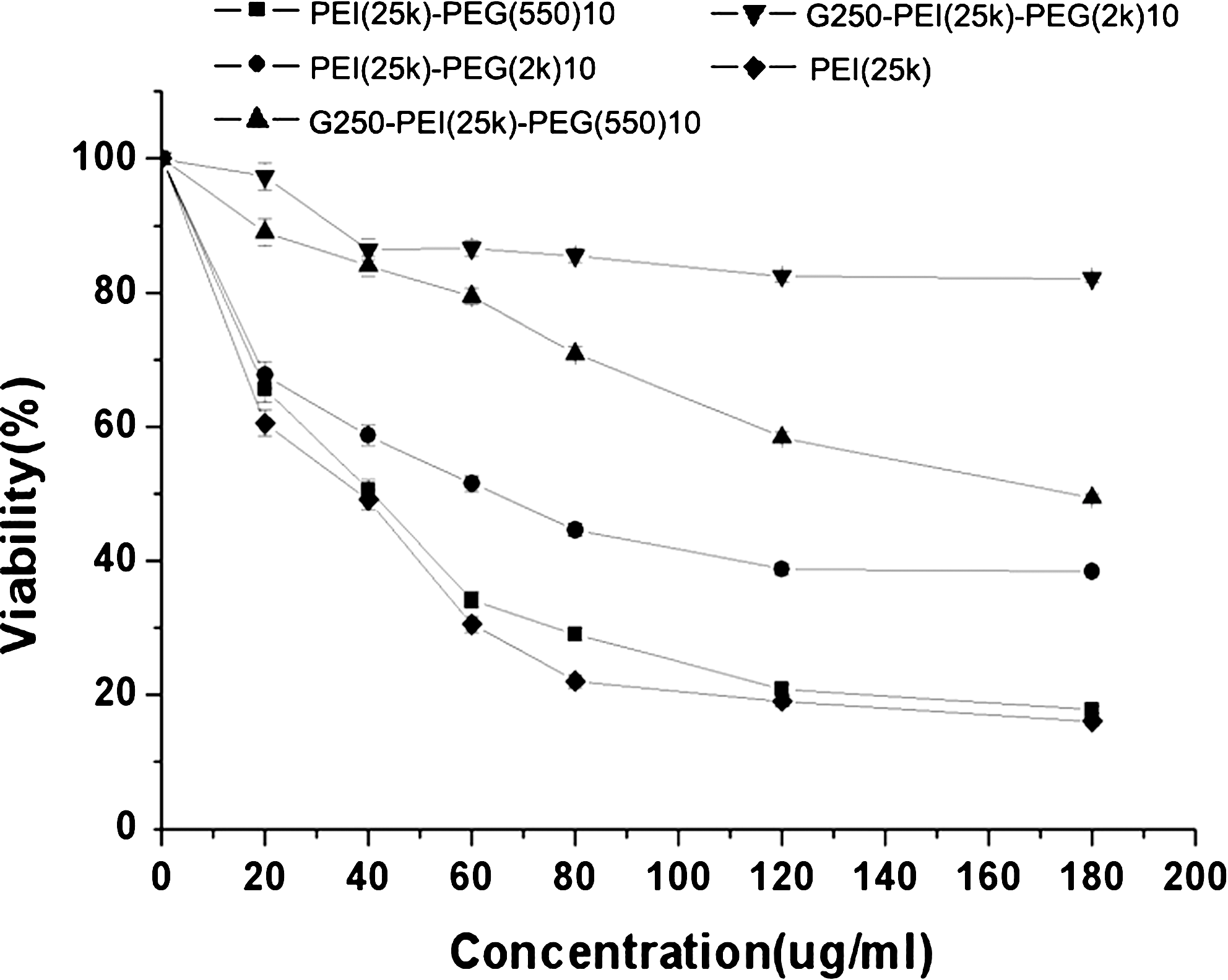
Cellular toxicity as determined by MTT assay. NIH/3T3 cells were incubated with PEI, PEI-PEG(550)10, PEI-PEG(2k)10, G250-PEI-PEG(550)10, or G250-PEI-PEG(2k)10 at various concentrations. The viability of nontreated cells was arbitrarily defined as 100%.
In vitro transfection
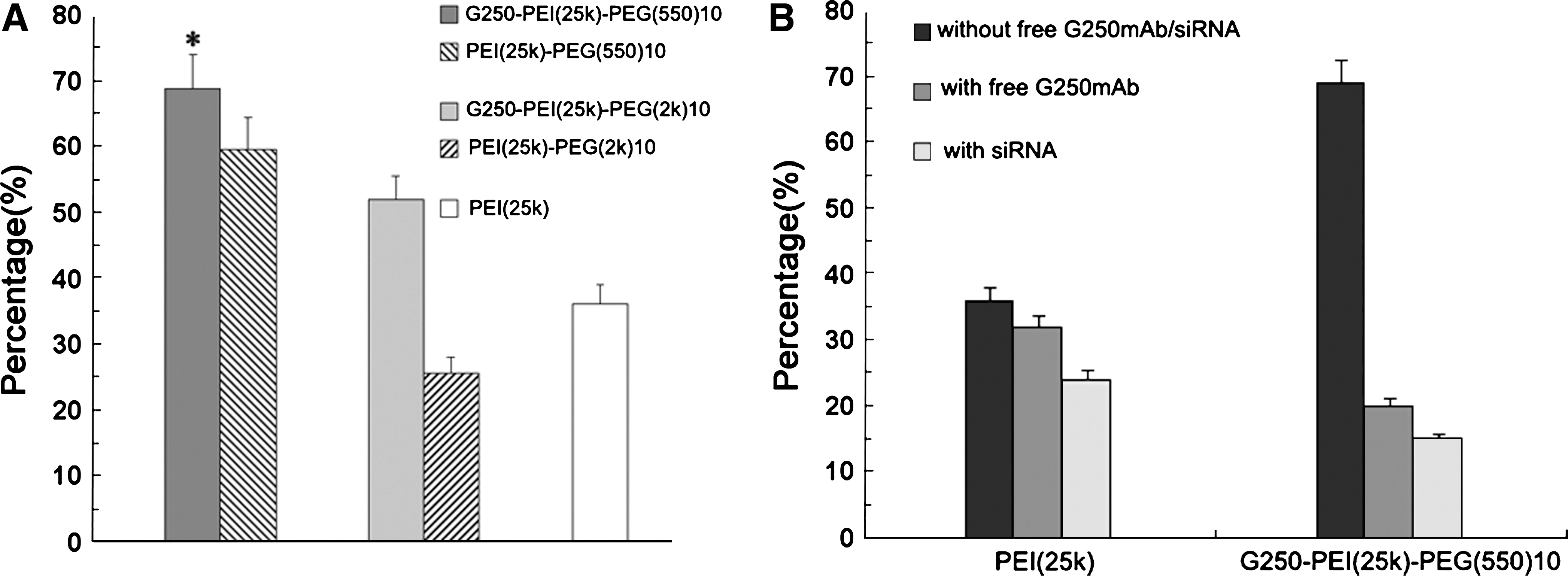
In vitro transfection was performed in the HeLa cell line, which expresses a high level of G250 antigen. Figure 5A shows the transfection efficiencies of PEI-PEG(550)10, PEI-PEG(2k)10, G250-PEI-PEG(550)10, G250-PEI-PEG(2k)10, and PEI. The highest transfection efficiency was seen with G250-PEI-PEG(550)10, that is, 68.9 ± 5%. The efficiency decreased to 30% after blocking the cells with free G250 mAb. Downregulating G250 expression by siRNA treatment further decreased expression to 15% (Fig. 5B). This result indicates the targeting effect of G250 antibody modification. To confirm the feasibility of G250-PEI-PEG(550)10 for in vivo gene delivery, transfections were done in the presence of 20 or 30% serum. The presence of serum decreased the transfection efficiency to various degrees for all the polymers. However, G250-PEI-PEG(550)10 maintained transfection efficiency up to 34.9 ± 1.8% in the presence of 30% serum, which was 2-fold higher than with the other polymers (Fig. 6).
(
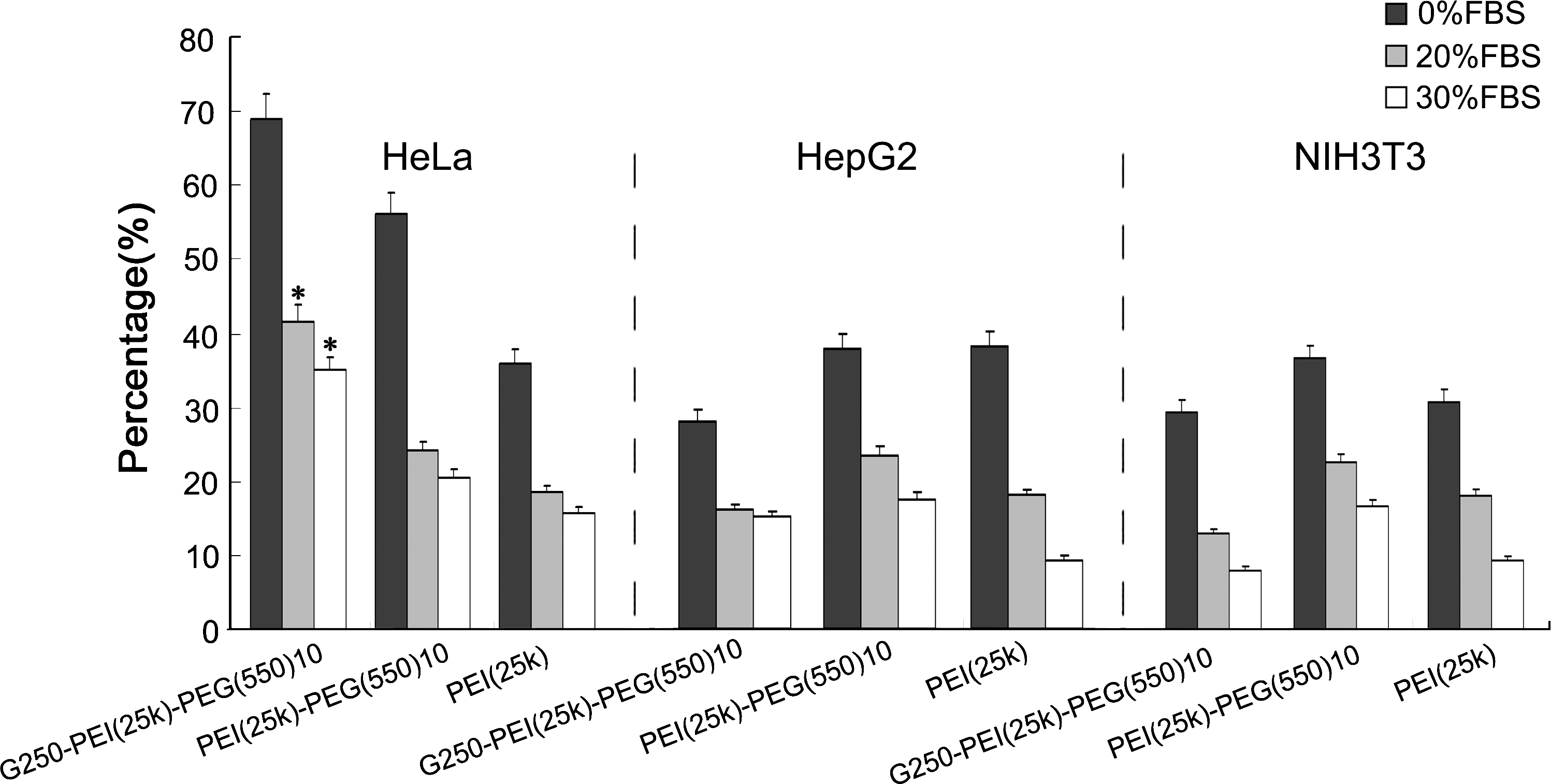
Transfection efficiency of PEI, PEI-PEG(550)10, and G250-PEI-PEG(550)10 as analyzed by FACS in the presence of 20% and 30% FBS in three cell lines. In HeLa cells the efficiency of transfection with G250-PEI-PEG(550)10 (41.7 ± 2 and 34.9 ± 1.1%) was significantly higher than with PEI-PEG(550)10 (24.2 ± 1.3 and 20.6 ± 1.1%) and PEI (18.4 ± 0.9 and 15.7 ± 0.8%) in the presence of 20 and 30% FBS, respectively (*p < 0.05). In HepG2 and NIH/3T3 cells, however, the presence of serum decreased the transfection efficiency to 10–25% for all three polymers.
In vivo transfection
G250-PEI-PEG(550)10, PEI-PEG(550)10, and PEI were used for the in vivo experiments. Figure 7A–C shows sections of GFP gene-transfected tumors, which express green fluorescence. GFP-positive cells were seen in both PEI- and G250-PEI-PEG(550)10-transfected tumors. Because it was difficult to count the number of positive cells, the luciferase assay was applied in this study. As shown in Fig. 8, the luciferase expression in G250-PEI-PEG(550)10-transfected tumors was 3.49 × 106 ± 0.3 × 106 RLU/mg proteins (n = 7), in comparison with 1.54 × 106 ± 0.2 × 106 RLU/mg proteins in the PEI group (n = 7). In all other organs, the expression of luciferase was lower than 0.22 × 106 RLU/mg proteins.

Images of GFP gene-transfected cells in tumor. (
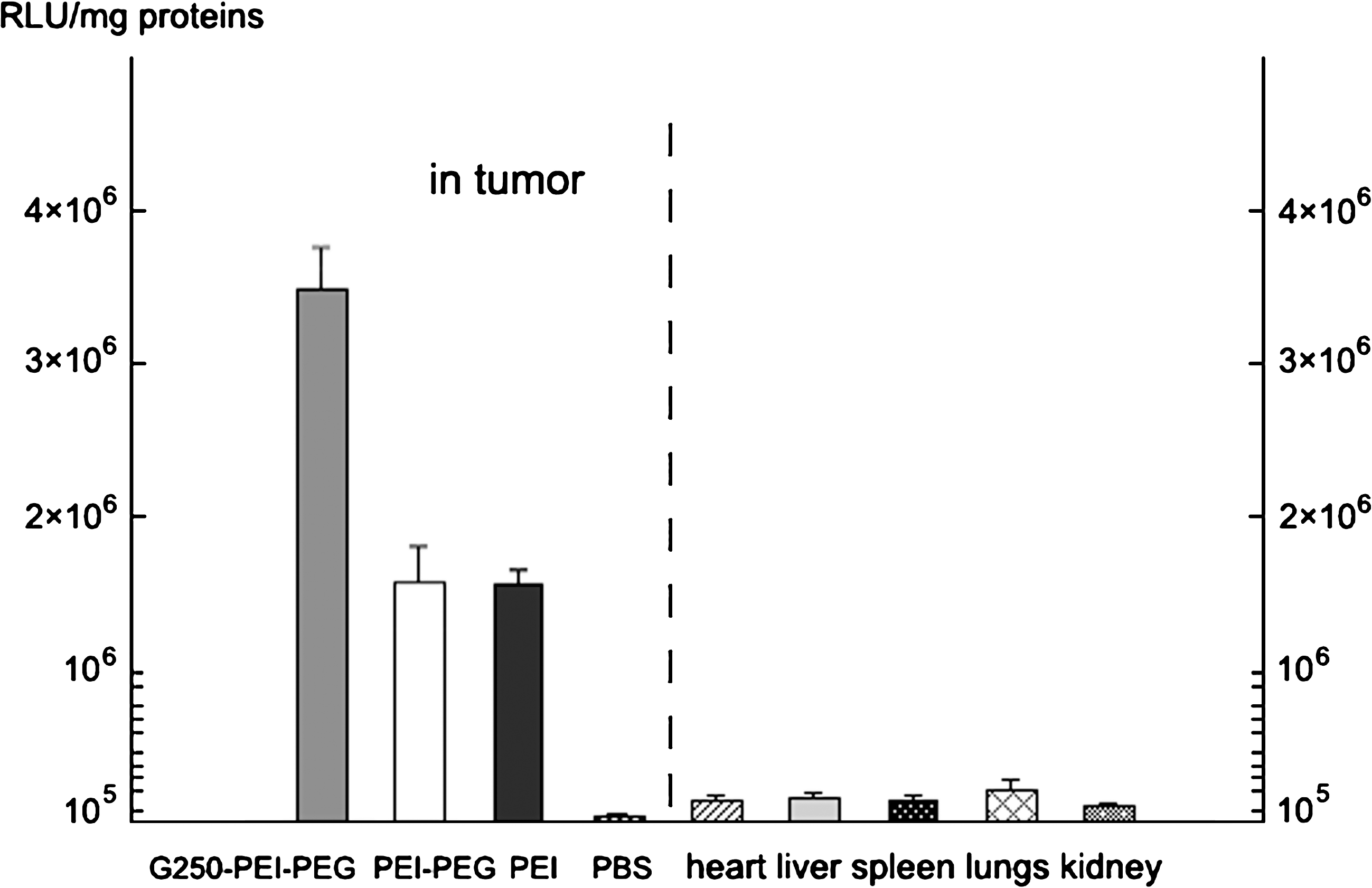
Luciferase expression in HeLa tumors and other organs in nude rats. Luciferase expression was (3.49 ± 0.3) × 106 RLU/mg proteins (n = 7) in G250-PEI-PEG(550)10-transfected tumors. However, it was (1.6 ± 0.3) × 106 and (1.54 ± 0.2) × 106 RLU/mg proteins in PEI-PEG(550)10- and PEI-transfected tumors, respectively (n = 7). Luciferase expression in other organs was low. *p < 0.05, G250-PEI-PEG(550)10 versus PEI.
Discussion
Because of its lack of toxicity, low immunogenicity, and antigenicity, PEG modification has been a common approach to improve the application of nonviral gene vectors (Dreborg et al., 1990; Petersen et al., 2002b). It is known that the molecular weight of PEG and the molecular ratio between PEG and polymers are important for gene delivery efficiency. The present study chose PEG of 550 Da and 2000 Da (2k).
Modification conditions were optimized by comparing various molecular weights of PEG and ratios of PEG to PEI. As shown in Figs. 1 and 2, modification with PEG(550) improved the transfection of PEI, and the optimal molecular ratios between PEG(550) and PEI were found to be 5 and 10. The structure was characterized by 1H NMR spectrum (Fig. S2,
A gel retardation assay was typically used to evaluate the gene vectors for the formation of DNA polyplexes. Figure 3 shows that when the weight ratio was 1, the mobility of DNA was completely retarded by both G250-PEI-PEG(550)10 and G250-PEI-PEG(2k)10. This suggested that these two conjugates can sufficiently condense DNA and form polyplexes. However, the size of the polyplexes, apart from the ability to form polyplexes, is critical for transfection efficiency. As shown in Figs. 1 and 2, efficient transfection required polymer-to-DNA ratios as high as 5 for PEI-PEG(550) and 50 for PEI-PEG(2k). This suggests that although polyplexes can form at low weight ratios, much higher weight ratios are required for the polymers to condense DNA into nanoparticles that can be easily taken up by cells.
After G250 conjugation, the viability of G250-PEI-PEG-treated cells was more than 80% with the concentration of polymers from 20 to 60 μg/ml. The transfection efficiency of G250-PEI-PEG(550)10 in HeLa cells was as high as 70%. A possible explanation could be that PEG modification reduced the toxicity, and G250 modification enhanced transfection by generating a targeting effect. In vitro transfections were also done in the presence of 20 and 30% FBS. The presence of serum decreased the transfection efficiency of all polymers. However, G250-PEI-PEG(550)10 maintained significant transfection efficiency (up to 35%) in the presence of 30% FBS. These findings suggest the feasibility of G250-PEI-PEG(550)10 for in vivo study.
In vivo experiments show that G250-PEI-PEG(550)10 was able to deliver pEGFP-C1 plasmid into tumors. Although green fluorescence was seen in both PEI- and G250-PEI-PEG(550)10-transfected tumors, luciferase expression in the G250-PEI-PEG(550)10 group was more than 2-fold that of PEI (Fig. 8). It indicates that G250 and PEG modification significantly enhanced in vivo transfection efficiency and gene expression. The level of luciferase protein, 3.49 × 106 ± 0.3 × 106 RLU/mg proteins, is comparable to that presented in a previous report (Goula et al., 1998). The expression of luciferase was much lower in other organs than in tumors, suggesting that G250 modification generated a tumor-targeting effect and significantly reduced nonspecific gene delivery. H&E staining did not show obvious damage to the liver, suggesting good biocompatibility of the G250-PEI-PEG conjugates (Fig. S1,
In summary, G250 mAb and PEG can be used to modify PEI. The G250-PEI-PEG(550)10 conjugate showed targeted gene delivery to HeLa cells in vitro.
PEG modification of PEI impaired the inhibitory effect of serum and significantly enhanced transfection efficiency in the presence of serum in vitro and in vivo. These results indicate that G250-PEI-PEG(550)10 conjugate may be a useful nonviral gene vector for cancer gene therapy.
Footnotes
Acknowledgments
This study was financially supported by grants from the National Outstanding Youth Fund (no. 30725030), the 973 Key Project (no. 2005CB623904), and the NSFC (nos. 20774050 and 30570385). The authors thank Professor Cunxian Song (Institute of Biomedical Engineering, Chinese Academy of Medical Sciences, Tianjin, China) for providing plasmid pEGFP-C1.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.