
Abstract

Introduction
More than two decades have passed since the initial technology was created to develop recombinant herpes simplex viruses (Post and Roizman, 1981; Roizman and Jenkins, 1985; Meignier et al., 1988). Since that time, herpes simplex viruses have been engineered into amenable tools for gene therapy. There are a wide range of herpes simplex virus (HSV)-based vector platforms and packaging strategies, which are continually being improved to fit the needs of a particular application. In this review, we present the biology of HSV, and explore various HSV-based vectors used in gene therapeutic approaches and the severity of their ensuing innate and adaptive immune responses.
Herpes Simplex Viruses
Structure
HSV type 1 and type 2 are closely related alphaherpesviruses, and part of the larger Herpesviridae family. The herpesviruses all have similar virion morphology: envelope, tegument, capsid, and core (Mettenleiter, 2003) (see Fig. 1). HSV virion structure has been imaged by a high-resolution technique, cryoelectron tomography, which determined the virion to be a spherical particle approximately 186 nm in diameter (Grunewald et al., 2003). HSV-1 and HSV-2 have linear double-stranded DNA genomes of 152 and 155 kb, respectively, which reside in the core (McGeoch et al., 1988; Dolan et al., 1998). The genome is composed of two parts, L (long) and S (short) unique regions, each flanked by inverted repeats (Wadsworth et al., 1975). Surrounding the core is an icosahedral capsid (Homa and Brown, 1997; Newcomb et al., 1999), which is itself coated with the tegument, a layer of at least 15 types of proteins. The outermost layer is a lipid envelope studded with 11 types of glycoproteins (Spear, 2004). The glycoproteins of the lipid envelope perform a variety of functions including viral entry, spreading, and immune evasion. Although all 11 do not appear to be absolutely necessary for a successful infection in vitro, gB, gD, gH, and gL are required (Spear, 1993, 2004; Steven and Spear, 1997).
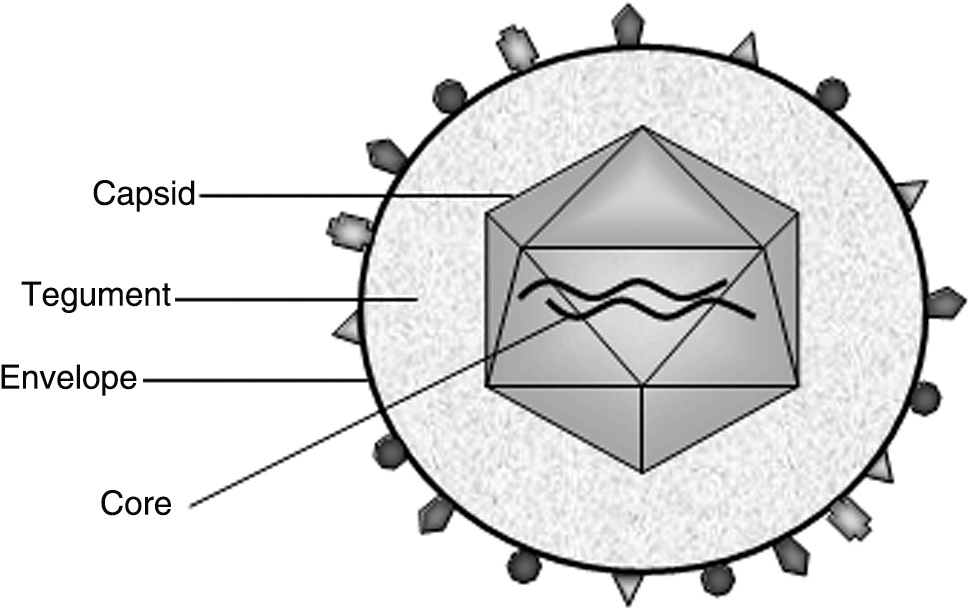
The key elements of HSV virion morphology include the envelope with associated glycoproteins, tegument, icosahedral capsid, and DNA-containing core.
Entry
Humans are the primary host for this naturally occurring neurotropic virus (Mettenleiter, 2003). In general, infection occurs when HSV enters the epithelial cells of the skin or mucous membrane. Initial association of the virion envelope with the host cell surface is mediated by the presence of cell surface glycosaminoglycans (GAGs), particularly heparan sulfate, which act as ligands for glycoproteins gB and gC (WuDunn and Spear, 1989; Herold et al., 1991; Shieh et al., 1992). The absence of this interaction does not prevent entry, but simply slows the process (Gruenheid et al., 1993; Tal-Singer et al., 1995). After the virion envelope attaches to the host cell, herpesvirus entry mediator A (HveA; formerly HVEM) and nectin-1 (formerly HveC) mediate the docking and uptake of HSV (Montgomery et al., 1996; Kwon et al., 1997; Geraghty et al., 1998; Warner et al., 1998). HveA is a member of the tumor necrosis factor (TNF)-α/nerve growth factor (NGF) receptor family, whereas nectin-1 is a member of the immunoglobulin superfamily (Montgomery et al., 1996; Geraghty et al., 1998). Although HveA and nectin-1 have no homology, they are both ligands for the viral glycoprotein gD, the virion entity responsible for the initiation of fusion (Geraghty et al., 1998; Nicola et al., 1998; Krummenacher et al., 1999). Viral envelope fusion with the cell membrane permits the delivery of the tegument proteins and the DNA, contained within the capsid, into the host cell cytoplasm. The capsid is transported via microtubules to the nucleus, where the genome enters via the nuclear pore complex and the linear DNA circularizes (Sodeik et al., 1997; Ojala et al., 2000).
Life cycle
After entry, the HSV life cycle can advance down one of two conduits: lytic or latent infection. During lytic, or productive, infection, a stereotyped temporal cascade of gene expression occurs. Along with the capsid, other tegument proteins, such as VP16, are also transported to the nucleus (Post et al., 1981). VP16 is responsible for initiating the transcription of immediate-early (IE) genes, which express the infected cell proteins ICP0, ICP4, ICP22, ICP27, and ICP47. Expression of the IE genes is required for the early and late genes; ICP4 and ICP27 are of particular importance, as we discuss later. Early and late gene expression generates the elements involved in viral DNA synthesis, replication, and the structural components of the virion (Frampton et al., 2005). The end result of a lytic infection is mature viral particles released from the host cell via exocytosis or cell lysis.
During latent infection the lytic transcription cascade, described previously, is blocked. Latency is typically established in sensory neurons whose terminals are adjacent to the infected epithelial cells. Virus enters the neurons and is transported to the nucleus, where it remains in a suppressed state. A set of RNAs termed the latency-associated transcripts (LATs) is produced, but remain untranslated (Croen et al., 1987; Stevens et al., 1987). Certain factors can stimulate reactivation of HSV at any time during latency. At that point, the lytic cascade of gene expression is no longer suppressed; viral transcription, DNA replication, and particle production and egress resume.
Herpes Simplex Viral Vectors
The use of virus-based vectors to deliver DNA to animal models of disease has become increasingly popular. Various therapies, which demonstrated promise in animals, have now progressed into clinical trials, where some are demonstrating early signs of success. Even these few cases of translation from basic research to patient therapy are encouraging. These successes promote the continued study and fine-tuning of viral vectors for use as tools for gene therapy. As more information is uncovered, it becomes apparent that certain vector platforms are inherently suited to treat particular diseases. HSV vector platforms are unique, compared with others available, because of their high efficiency of gene transfer, ability to establish long periods of latency, extensive host cell range, and potentially large transgene capacity. In addition, the HSV vector remains as an extrachromosomal episome, which decreases the likelihood of an insertional mutation in the host's genome (Mellerick and Fraser, 1987). The ability of HSV to infect and express transgenes from neurons (a nondividing cell type) also makes it ideal for the design of gene therapies that target diseases of the CNS. Studies that have mutated portions of the HSV genome have shown that only half of the open reading frames (ORFs) are required for viral replication in vitro. This enables their replacement with relatively large genes, multiple genes, or regions including regulatory elements and promoters. The disadvantages of this vector system, such as cytotoxicity and short duration of transgene expression, have become obstacles that may be overcome with further engineering. HSV-1 is the most commonly used herpesvirus for gene transfer; therefore we concentrate on it. Subsequent discussion focuses on the two types of HSV vector: recombinants and amplicons. It is important to review both types because of the different responses they elicit from the host immune system.
Recombinants
Recombinant HSV (rHSV) vectors are generated by targeting the insertion of a gene of interest into a locus occupied by an HSV wild-type gene, using homologous recombination. As mentioned previously, extensive regions of the wild-type HSV-1 genome can be deleted or replaced without disrupting the viral life cycle. Insertion of a transgene into one of these unessential stretches would produce a replication-competent rHSV vector. Insertion of a heterologous gene into an essential region yields a replication-defective vector. Replicating vectors have restricted utility, such as cancer therapies, in which viral replication is limited to dividing tumor cells (oncolytic vectors). The cytolytic activity produces the toxicity that achieves the objective of destroying the tumor cells. However, in applications in which extended periods of gene expression are desired, viral replication incites large amounts of cytotoxicity from viral protein synthesis and death of cell types that permit replication. Therefore, replication-defective rHSV vectors are the more widely used type. The existence of two IE genes, ICP4 and ICP27, is imperative for viral replication. Studies using either deletion mutant have discovered that ICP4 and ICP27 are potent transcriptional trans-activators for expression of other early and late viral genes (DeLuca et al., 1985; Sacks et al., 1985). Accordingly, several replication-defective rHSV vectors are based on mutations in these two IE genes. Replication and virion packaging with these vectors is accomplished with complementing eukaryotic cell lines that provide the missing gene product in trans (DeLuca et al., 1985; Marconi et al., 1996).
rHSV vectors boast a transgene capacity of approximately 30 kb. Despite blocking viral replication, cytopathic effects are still observed with the use of these replication-defective HSV recombinant vectors. It appears that the nondeleted IE gene products are a source of toxicity (P.A. Johnson et al., 1992b). rHSV-1 vectors containing multiple IE gene deletions have diminished levels of toxicity, yet the consequence is a severe reduction in transgene expression (Krisky et al., 1998; Samaniego et al., 1998). These shortcomings of the rHSV vector systems have motivated the generation and use of an alternative HSV vector system, the HSV amplicon.
Amplicons
Compared with the rHSV vector, the HSV amplicon is a stripped-down vector containing far fewer viral genes. In 1982, Spaete and Frenkel created a “cloning–amplifying” vector, or the amplicon, from multiple repeats of defective HSV genomes (Spaete and Frenkel, 1982). HSV amplicons are eukaryotic expression vectors composed of two virally derived constituents, the HSV origin of DNA replication (OriS) and the cleavage/packaging signals (pac), as well as the transgene. Virions are packaged with HSV-1 helper virus. The DNA is replicated via a rolling-circle mechanism generating concatemers of the amplicon DNA, which is cleaved at the pac sequence. These DNA sequences, of approximately 152 kb, are packaged into the capsid.
Packaging
Packaging amplicon DNA into viral particles requires helper virus function to supply the machinery for DNA replication and the structural proteins of the virion. The role of helper virus was initially played by deletion mutants of HSV-1, which rendered them replication-defective rHSV (Geller et al., 1990). In these instances packaging occurred in a cell line providing the missing gene product. The result was a combination of amplicon and helper viral particles produced at certain ratios depending on the procedure employed; the goal in fine-tuning packaging procedures was to attempt to obtain a lower number of helper viral particles. High titers (108–109 infectious vector particles per milliliter) are obtained by helper virus packaging, yet several drawbacks exist. Through homologous recombination with the packaging cell DNA, the helper virus genome can revert to wild-type HSV at a relatively high rate (10–4) (Geller et al., 1990). This can be partially remedied by using complementing cells with shorter regions of the deleted gene, making homologous recombination less likely (10–6) (DeLuca and Schaffer, 1987; P.A. Johnson et al., 1992a). Wild-type HSV revertants aside, contaminating helper virus expresses low levels of gene products, causing cytopathic effects. The viral products can undergo traditional antigenic processing within the host, resulting in an immune response, which will likely decrease the levels of transgene expression.
Helper virus-free packaging has been developed as an alternative for packaging of HSV amplicon. The helper virus genome is supplied in a set of five overlapping, but distinct, cosmids or bacterial artificial chromosomes (BACs), in which the pac signals are inactivated through mutation (Fraefel et al., 1996; Saeki et al., 1998, 2001; Stavropoulos and Strathdee, 1998). These five elements and the amplicon DNA are cotransfected into permissive eukaryotic cells. Vector stock preparations are devoid of helper virus, and thus cause negligible cytopathic effects on transducing cells. The resultant titers range from 107 to 108 expressing viral particles per milliliter. A number of different sources stimulate immune responses in applications using HSV vectors. We explore more on immunity in the remaining sections.
Innate Immune Response
Innate immunity is the host's initial resistance to infection by microorganisms. The innate immune system typically recognizes common elements on the surface of invading pathogens. Several components of the innate immune system work together to destroy microorganisms; if they are not eliminated, innate immunity controls the infection and aids in initiating a proper adaptive immune response, which can take between 4 and 7 days. Wild-type HSV-1 activates the complement cascade (Da Costa et al., 1999). In addition, various immune cells are recruited and activated through the coordinated secretion of an assortment of cytokines and chemokines (Melchjorsen et al., 2006); these include the activation of macrophages, natural killer (NK) cells, dendritic cells (DCs), and γδ T cells (R.M. Johnson et al., 1992; Kawamura et al., 2006). Therapeutically delivered HSV-based vectors may contain elements that stimulate these responses; depending on the application, this may or may not be desirable.
Toll-like receptors and HSV glycoproteins
Toll-like receptors (TLRs) are part of the innate immune response and were initially thought to function predominantly against invading bacteria and fungi as pattern recognition receptors. However, many viruses have now been shown to activate the TLRs and stimulate an immune response. The TLR family consists of 10 different transmembrane proteins. They bind their ligands and subsequently signal through translocation of NF-κB to the nucleus. In the nucleus, pro- or antiinflammatory cytokine gene transcription is activated. HSV has been shown to bind TLR2 and TLR9 (Lund et al., 2003; Krug et al., 2004; Kurt-Jones et al., 2004). Viruses traditionally stimulate type 1 interferon (IFN) responses, which involve both chemokines activating inflammatory cells and cytokines causing helper T cell type 1 (Th1) lymphocyte differentiation. TLR9, on the surface of plasmacytoid and splenic DCs, has been shown to bind HSV double-stranded DNA (dsDNA), eliciting the production of IFN-α and interleukin (IL)-12 (Lund et al., 2003; Krug et al., 2004). HSV is also capable of interacting with TLR2 on the surface of macrophages and DCs. A set of proinflammatory cytokines, including TNF-α, IL-1β, and IL-12, is produced when HSV is permitted to activate TLR2 (Kurt-Jones et al., 2004; Aravalli et al., 2005). The TLR2-mediated response may not be protective, but pathogenic, leading to deadly encephalitis (Kurt-Jones et al., 2004). Fragments of HSV glycoprotein gD have induced an IFN-α response comparable to that of intact HSV (Ankel et al., 1998), which may imply that gD is a binding partner for a TLR. Some viral particle components of wild-type HSV (dsDNA and gD), which activate innate immunity, may be present when using rHSV or amplicons for therapeutic applications, and therefore these type 1 IFN responses should be anticipated.
Host consequences
Through evolution wild-type HSV has developed immune evasion mechanisms, which make it a formidable pathogen for the host's immune system. HSV employs means to inhibit humoral immunity and impede correct antigenic processing and presentation. It is important to review these viral strategies of immune evasion to understand whether their inclusion within an HSV vector system would be beneficial or detrimental to a particular application. HSV uses two independent mechanisms to inhibit the complement cascade. The first involves masking of antibodies. HSV glycoproteins gE and gI join in a complex to bind the Fc domain of IgG (Dowler and Veltri, 1984). Antibodies, which have bound infected cells, can no longer carry out their effector functions mediated through the Fc domain, including the binding of complement constituent C1q to the Fc. Similarly, viral glycoprotein gC binds to complement component C3, impeding C3 from being cleaved by the C3 convertase, whose products would coat the pathogen surface and initiate local inflammation (Lubinski et al., 1998).
HSV cleverly conceals itself from detection by cytotoxic T lymphocytes (CTLs), which would kill HSV-infected cells. A tegument protein termed virion host shutoff (vhs) has mRNase activity, effectively shutting down host cell protein synthesis. Along with other proteins, MHC class I molecules are not synthesized to present HSV components on the cell's surface. Not only are MHC I molecules synthesized in extremely low numbers, another mechanism of immune evasion causes their misfolding and retention in the endoplasmic reticulum (ER). An IE gene product, ICP47, binds and obstructs the function of TAP (transporter associated with antigen presentation), thus disabling CTLs from recognizing cells infected with HSV (Tomazin et al., 1996).
Inferring immune consequences to HSV vectors via wild-type HSV infections is one approach to determining their utility. A second strategy is to employ animal models to establish the efficacy and types of immune responses that will ensue from using a particular HSV-based vector.
Influence on vector utility
HSV-based vectors have been used in a variety of diverse applications, including gene transfer, as oncolytic viruses to destroy tumor cells, or as a vaccination platform. In the case of gene transfer, a known missing or mutated gene product that results in disease is replaced through delivery of a transgene, using the HSV vector. An immune response against the vector or the transgene would be detrimental to sustained transgene expression, undermining the efficacy of the therapy. Alternatively, when using HSV as a vaccination platform the preferred outcome is a directed immune response to the transgene. Costimulatory immune molecules or even the viral particle itself can act to stimulate the immune system to respond in a certain way. The desired results of these two applications are essentially opposites and would require that different HSV-based vectors and packaging strategies be used. For example, some of the natural mechanisms of wild-type HSV immune evasion, described previously, would be functional in gene transfer applications.
Recombinant HSV vectors and amplicons cause differential immune responses (see Table 1). The nondeleted IE gene products are a source of toxicity in applications using rHSV (P.A. Johnson et al., 1992b). Applications that employ HSV amplicon and helper virus-free packaging techniques do not encounter this issue because viral gene transcription does not occur, due to the absence of helper virus. rHSV-1 vectors containing multiple IE gene deletions have diminished levels of toxicity, yet the consequence is a severe reduction in transgene expression (Krisky et al., 1998; Samaniego et al., 1998). The virion itself, which is present independent of platform use, must contain the essential glycoproteins. Glycoprotein gD, which induces an IFN-α response, is one of the HSV glycoproteins required for successful viral transduction. This appears to be an inevitable response.
The neurotropic nature of HSV vectors has resulted in their frequent use in the CNS; thus, the brain has been a focal point in the study of HSV-generated immune responses. The first study to evaluate inflammatory responses in the murine CNS used an HSV amplicon expressing β-galactosidase packaged by a conventional helper virus-based strategy (Wood et al., 1994). Significant microglia activation and MHC class I upregulation was present from 2 days postinfection. MHC class II-positive cells, macrophages, and T lymphocytes were pervasive by day 4 and persisted (Wood et al., 1994). In another study, HSV amplicon, expressing β-galactosidase, was packaged by either a helper virus-based or helper virus-free technique and stereotactically injected into the CNS of mice. Five days after injection, after initial inflammation from the injection had resolved, mRNA levels of proinflammatory cytokines (IL-1β, TNF-α, and IFN-γ) and chemokines (monocyte chemotactic protein [MCP]-1 and interferon-γ-inducible protein [IP]-10), and of a cell adhesion molecule (intercellular adhesion molecule [ICAM]-1), were elevated in the mice receiving helper virus-based injections. These molecules had returned to near baseline levels in the mice given helper virus-free amplicon injections (Olschowka et al., 2003). This study demonstrated the role of contaminating helper virus in the CNS immune response. It appears that the mode of packaging greatly influences the extent of inflammatory response.
Adaptive Immune Response
In contrast to an innate response, adaptive responses are mediate by immune cells specific for an antigen. The clonal selection of lymphocytes gives the immune system the capacity to recognize and have memory for specific pathogens and to mount a rapid and potent response against subsequent attacks from that pathogen. Lymphocytes are the effector cells of adaptive immunity including B cells, which function in humoral immunity, and T cells, which are the mediators of cellular responses.
Antigen-presenting cells (APCs), such as DCs, take up pathogens, become activated, and migrate to the lymph nodes. In the lymph nodes they interact with T cells. The initial T cell–APC interaction (T cell priming) causes the release of various cytokines and chemokines, the identity of which determines whether naive CD4+ T cells will develop into T cell helper type 1 (Th1) cells or T cell helper type 2 (Th2) cells. Th1-type responses rely on the presence of the cytokines IFN-γ and IL-1, whereas Th2 responses occur in the presence of IL-4 and IL-10. Typically, the Th2 pathway will elicit a humoral response characteristically producing antibodies of the IgG1 isotype and are not inflammatory in nature. Alternatively, responses that proceed down a Th1 pathway typically produce antibodies of IgG2b isotype, and activate CTLs and a host of proinflammatory cytokines. Usually there is a combination of both of these responses. HSV-based vectors can be used to drive an adaptive immune response to a specific antigen and skew that response toward either a humoral or cellular response.
Vectors as vaccination platforms
Classically, vaccines have been of two kinds: (1) inactivated viruses, which generate a CD4+ T cell humoral response, and (2) live attenuated viruses that undergo minimal replication and induce CD8+ and CD4+ cellular and humoral responses. These two strategies, however, do not produce an effective vaccine for all microorganisms. In addition, some investigators are developing immune therapies for diseases that are targeted toward a pathogenic self-antigen, such as a misfolded or mutated protein. In these instances recombinant viral vectors can be used to express heterologous antigens to induce an immune response. The first report of an attempt to design a vaccine using an HSV-based vector was by Roizman and colleagues in 1984. The gene encoding the hepatitis B virus (HBV) surface antigen was inserted into the thymidine kinase locus of HSV-1. Infected cells successfully expressed the HBV surface antigen under the control of endogenous HSV promoters and regulatory regions (Shih et al., 1984). The large transgene capacity of HSV-based vectors is advantageous as a vaccination platform. It permits the codelivery of the transgene (antigen of interest) as well as of other genes that may act to shape or direct the immune response (see the next section).
Immune shaping
Cytokines present in the environment at the time of T cell priming help to establish the pathway down which the immune response will proceed. Numerous vaccination strategies attempt to influence or direct the immune response, toward either a Th1 type or Th2 type; the presence of particular cytokines is one way to accomplish this goal. The presence of IFN-γ and IL-1 will push the development of a Th1-type response. This type of response is useful when cellular immunity and inflammation are desired. The presence of IL-4 and IL-10 will skew a response toward the Th2 type. These cytokines bring about a strong humoral response, avoiding the often unnecessary, and dangerous, inflammation and presence of CTLs. The preferred cytokine gene can be inserted into the HSV-based vector, allowing for its coexpression with the antigen of interest, increasing the likelihood that a particular response is established. Another consideration when attempting to direct an adaptive response is the choice of transgene within the vector. Whole gene products may contain several different epitopes, which can be T or B cell epitopes. Expressing only a portion of an antigen of interest, including a specific epitope, or coexpressing an epitope that is a strong inducer of a known response type, can push the response toward one that is more humoral or cellular in nature. Examples of this are described in the next section.
Applications
Numerous preclinical studies have employed HSV vector systems to deliver transgenes to animal models of disease with the goal of inducing an adaptive immune response or providing a therapeutic transgene; some examples include Alzheimer's disease, chronic lymphocytic leukemia, and prostate cancer.
As the leading cause of dementia in the United States, Alzheimer's disease (AD) is widely studied, and many attempts have been made to develop therapies for its sufferers. Increasing genetic and biochemical evidence has implicated a cleavage product of amyloid precursor protein (APP), termed amyloid-β (Aβ), as having a lead role in the pathogenesis of AD. After cleavage, Aβ undergoes assembly into aggregated forms. These small soluble aggregates, termed Aβ oligomers, are postulated to compromise synaptic function (Lacor et al., 2004; Lesne et al., 2006), which will lead to synapse loss and neuronal death. Some therapeutic approaches have attempted to raise an immune response to Aβ by traditional vaccination paradigms. After several animal studies found encouraging results in their attempts to employ anti-Aβ antibodies, without unfavorable responses, a human clinical trail was initiated. Using Aβ42 fibrillar peptide and an adjuvant, QS-21, AD patients were immunized. The vaccination stimulated the production of anti-Aβ antibodies and appeared to slow disease progression in many subjects (Hock et al., 2003). Yet, severe brain inflammation occurred in a subset, approximately 6%, of the patients and the trial was ended. Patient data suggested the inflammatory response may have been due to the presence of infiltrating T cells (Nicoll et al., 2003). Since this trial one goal of AD therapy has been to produce a potent humoral response, while avoiding cytotoxic and inflammatory T cell activation; gene therapy could accomplish this by directing an adaptive immune response to Aβ.
Bowers and colleagues peripherally vaccinated transgenic Tg 2576 AD mice, or control nontransgenic mice, with an HSV amplicon vector containing a transgene for Aβ1–42 (HSVAβ) alone or with Aβ1–42 fused in frame with tetanus toxin fragment C (HSVAβ/TtxFC) (Bowers et al., 2005). As Aβ is an endogenous protein, self-tolerance exists and anti-Aβ antibodies are not produced under normal conditions. To raise antibodies to Aβ self-tolerance must be broken, using techniques such as overexpression and/or pairing with a known molecular adjuvant, such as TtxFC. Both transgenes delivered by HSV amplicon vector produced excellent antibody titers; yet, HSVAβ–TtxFC generated IgG1 isotypes indicative of a Th2 cellular response, whereas HSVAβ produced first IgM, then IgA isotypes. Four of six AD mice that received a second subcutaneous HSVAβ injection became ataxic and died within 1 week. Analysis by RT-PCR demonstrated upregulation of proinflammatory transcripts in the hippocampi, yet there were no significant increases in proliferating T cells or activated microglia. Nontransgenic mice receiving injections did not experience these phenomena. A reduction in the area and the number of Aβ plaques was shown in AD mice receiving the molecular adjuvant HSVAβ–TtxFc, as well as in the surviving HSVAβ recipients. These data highlight the importance of directing the immune response in an appropriate manner. The strong B cell epitope contained within TtxFC is likely responsible for the Th2-type response seen in the mice receiving this vector.
Summary
HSV-1 vectors of both the recombinant and amplicon types have been developed as efficient transfer vehicles. Although epithelia and neurons are the natural cellular targets for wild-type HSV infection, studies in a number of laboratories have demonstrated that a number of cell types can support transduction. Because the amplicon does not encode any HSV genes it has been exploited as a vaccine platform. Studies in Alzheimer's disease mouse models indicate that the amplicon vaccine platform can be exploited to bias an immune response. Immune shaping has been achieved by the inclusion of specific epitopes into immunogens that drive Th2-type immune responses. These data portend the progressive development of the amplicon as a vaccine platform that may in the future have clinical utility.
Footnotes
Acknowledgments
This work was supported by NIH RO1-AG020204 (H.J.F.).
Author Disclosure Statement
Deborah Ryan has no conflicts of interest to declare. Howard Federoff is a founder and scientific advisory board member for a biotech start-up, MedGenesis Therapeutix, which had previously had an option to Amplicon Technologies.