
Abstract
Liver-based gene therapy approaches demonstrated that high-capacity adenoviral vectors (HC-AdVs) can persist life-long in mice and for 2 years or longer in rats, dogs, and nonhuman primates. However, the molecular status of episomal HC-AdV DNA molecules and the mechanism of vector genome maintenance have not been analyzed. HC-AdV lacks all viral coding sequences including early gene region 4 (E4), which prevents concatemerization in wild-type adenovirus. Therefore, we addressed whether concatemerization or circularization of HC-AdV DNA occurs in transduced cells. We employed pulsed-field gel electrophoresis and a sensitive concatemer/circle-specific polymerase chain reaction (PCR). To test for replication as a potential mechanism for maintenance, we developed a methylase/restriction endonuclease-based system using methylation-marked HC-AdV. We found that unlike ΔE4 mutant virus, only monomers of HC-AdV genomes were observable in vitro. Using our methylase/restriction endonuclease-based system, no replication of HC-AdV was sensed in various cell lines. However, concatemer formation of HC-AdV could be induced after coinfection with an E4-deleted helper virus, indicating that linkage of genomes may be supported by replication. To examine HC-AdV DNA molecules in vivo, C57BL/6 mice were injected and vector DNA in liver was analyzed. In concordance with our in vitro results, exclusively linear monomers were detected. To sense the replication status of HC-AdV genomes, we established a sensitive real-time PCR. Our results indicated that the input transduced DNA genomes were the persistent molecules in murine liver. In summary, we demonstrated that HC-AdV genomes persist predominantly as replication-defective monomeric genomes.
Introduction
HC-AdVs have been shown to result in long-term phenotypic correction in various animal models covering various disease models. In multiple liver-based gene transfer studies we and others have shown that after a single injection nonintegrative HC-AdVs are maintained lifelong in mice and for up to 2 years in rats (Morral et al., 1998; Schiedner et al., 1998; Kim et al., 2001; Ehrhardt and Kay, 2002; Toietta et al., 2005). Also in dogs and nonhuman primates, HC-AdV transduction of the liver can lead to long-term HC-AdV genome persistence and transgene expression (Morral et al., 1999; Brunetti-Pierri et al., 2007). To date, the longest period of transgene expression (up to 964 days) was observed by Brunetti-Pierri and colleagues using an HC-AdV for hepatic transduction of baboons (Brunetti-Pierri et al., 2009). Importantly, it was demonstrated that hepatic transduction is not accompanied by chronic toxicity because of the absence of viral gene expression (Morral et al., 1998, 1999; Brunetti-Pierri et al., 2007). However, the molecular status of episomal HC-AdV DNA molecules and the mechanism of vector genome maintenance have not been established. We speculated that several molecular forms or mechanisms such as vector genome concatemer formation, circular genome formation, and/or replication may potentially be involved in persistence.
The HC-AdV genome contains the adenoviral inverted terminal repeats (ITRs) and a packaging signal at the 5′ end. Foreign DNA of up to 36 kb including the therapeutic transgene or stuffer DNA with potentially stabilizing effects can be inserted. Several HC-AdV genomes with different stuffer DNAs have been developed and duration of transgene expression was dependent on the DNA sequences contained in the adenoviral vector (Parks et al., 1999). Our previously published HC-AdV contains a matrix attachment region of the murine immunoglobulin κ locus and alphoid repeat DNA sequences from a centromeric region on human chromosome 17 (Ehrhardt and Kay, 2002).
Similar to linear DNA and recombinant adeno-associated virus (rAAV), the HC-AdV genome molecule enters the host cell as a linear DNA molecule. It is established that in target cells, which were transduced with either linear naked DNA or rAAV, vector DNA concatemer formation takes place (Leahy et al., 1997; Miao et al., 1998; Nakai et al., 2000; Chen et al., 2003). After transduction with linear nonviral DNA, these concatemers are believed to provide increased transgene expression levels in vitro and sustained transgene expression in vivo (Leahy et al., 1997; Chen et al., 2003). Upon infection with linear rAAV genomes, established double-stranded DNA (dsDNA) molecules serve as substrates for rAAV genome recombinations at the AAV ITRs. This process is believed to be mediated by the cellular DNA repair machinery. It results in circularization and concatemerization of episomal rAAV vector genomes. Altogether, these studies indicate that transgene expression levels and duration of the vector DNA in transduced cells may be influenced by the DNA molecular structure of the transduced vector.
Because of the linear nature of the adenoviral genome, it represents a potential substrate for concatemerization or circularization by the double-strand break repair (DSBR) machinery of infected cells. After infection of eukaryotic cells with wild-type adenovirus serotype 5 (wtAd5), the adenoviral early E4 11-kDa protein, encoded by E4 open reading frame 3 (E4 ORF3), and the adenoviral E4 34-kDa protein, encoded by E4 ORF6, form a complex with and inactivate key components of the DSBR system (Stracker et al., 2002, 2005; Carson et al., 2003; Weitzman, 2005). This prevents concatemer formation during wtAd5 infection. However, during infection with E4 ORF3/6 double mutants (ΔE4 mutant), the viral genomes are linked to one another at the termini, leading to head-to-head, tail-to-tail, and head-to-tail junctions (Weiden and Ginsberg, 1994; Jayaram and Bridge, 2005). In the present study we investigated whether HC-AdVs lacking all viral coding sequences including E4 form concatemers or circular molecular structures. Because of the lack of E4 in the genome of HC-AdV the DSBR pathways should not be inhibited.
To analyze whether HC-AdV genome replication independent of adenoviral early genes E1 and E2 takes place and potentially contributes to persistence of vector DNA, we established a methylase/restriction endonuclease-based system. A similar approach was also used by Nelson and Kay to determine the replicative status of first-generation E1-deleted adenoviral vectors (Nelson and Kay, 1997). We generated producer cells for amplification of methylation-marked HC-AdV by addition of a methyl moiety by methylase PaeR7 onto the N6 position of the adenine base of XhoI sites (Gingeras and Brooks, 1983). In this system, viral replication restores XhoI cleavage by removal of methylation. Thus, XhoI cleavage indicates replication of HC-AdV genomes.
In the present study we found that in contrast to rAAV vectors and linear nonviral DNA there was no linkage of recombinant adenoviral DNA molecules on infection of target cells. We provide evidence that nonreplicative linear monomeric genomes are responsible for persistence of transgene expression from HC-AdV in vitro and in vivo.
Materials and Methods
Cell culture and viruses
Human embryonic kidney 293 cells (CRL-1573; American Type Culture Collection [ATCC], Manassas, VA) and HeLa cells were grown in minimal essential medium (MEM; Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (FBS; Invitrogen). Hepatocyte-derived HuH-7, SK-HEP-1 (ATCC HTB-52), and murine Hepa 1A cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 10% FBS and 0.1 mM nonessential amino acids (Invitrogen). 293N3S cells, a 293 derivative capable of both adherent and suspension growth, are the ancestor cells of the HC-AdV producer cell line 116 (Palmer and Ng, 2003). This cell line carries a hygromycin resistance gene, expresses Cre recombinase, and was grown in MEM supplemented with 10% FBS (Sigma-Aldrich, Taufkirchen, Germany) and hygromycin B (100 μg/ml; Invitrogen). W162 cells (ATCC CRL-2783), a Vero cell derivative stably expressing the E4 region of Ad5, supports growth of adenoviral E4 deletion mutants. This cell line was maintained in MEM with 2 mM
Large-scale HC-AdV production was performed in 116 cells as described earlier (Palmer and Ng, 2003; Jager et al., 2009). In brief, 10 confluent 15-cm dishes of 116 cells were transferred into 1 liter of MEM supplemented with 10% FBS and hygromycin B (100 μg/ml). Over the following days, the same growth medium was added to a final volume of 3 liters (500 ml on days 2 and 3, 1000 ml on day 4). On day 5, 3 liters of 116 cells at 3 × 105 to 4 × 105 cells/ml were harvested by centrifugation, resuspended in 5% volume medium, and coinfected with 100% of the crude lysate from one 15-cm dish of serial passaging step 3 and AdNG163R-2 helper virus (Palmer and Ng, 2003) at 1 infectious unit per cell. Virus adsorption was performed at 37°C on a magnetic stir plate for 2 hr, after which medium (MEM supplemented with 5% FBS) was added to a final volume of 2 liters. Coinfected cells were harvested 48 hr later for lysis and HC-AdV virions were purified by one step and one continuous CsCl ultracentrifugations. In an alternative simplified protocol, purified HC-AdV virus (obtained by the method described previously) instead of crude lysate was used as inoculum at a dose of 100 infectious units per cell for coinfection of 3 liters of 116 cells.
HC-AdV virus FTC-hFIX-lucRNAi (Rauschhuber et al., 2008) was based on the parental plasmid pAdFTC (Ehrhardt and Kay, 2002) and contains adenovirus serotype 5 ITRs, the packaging signal, the matrix attachment region of the murine immunoglobulin κ locus and alphoid repeat DNA sequences from human chromosome 17. The human coagulation factor IX (hFIX) expression cassette was described previously (Miao et al., 2000) and contains two liver-specific enhancers (hepatic control region and the apolipoprotein A enhancer) and a human α1-antitrypsin promoter controlling the expression of the hFIX minigene. In addition, this HC-AdV contains an expression cassette encoding small hairpin RNA (shRNA) against firefly luciferase driven by the human U6 small nuclear RNA (snRNA) promoter.
Adenovirus serotype 5 ΔE4 mutant (H5dl1004, lacks nucleotides 2845 to 981 of E4, which equals ORF2-ORF7 of the E4 coding sequence; Sandler and Ketner, 1989) was amplified in W162 trans-complementing cells (Bridge and Ketner, 1989; Stracker et al., 2005) and wtAd5 was amplified in 293 cells.
Purified virions, prepared by CsCl equilibrium density centrifugation, were used for all experiments. Transduced cells were harvested with trypsin and were washed with phosphate-buffered saline (PBS) before DNA preparation to remove any uninfectious viral particles. For pulsed-field gel electrophoresis experiments, HC-AdV virus was used at doses from 3600 viral particles (VP)/cell up to 15000 VP/cell, wtAd5 at a dose of 10 VP/cell, and ΔE4 virus at a dose of 3 to 400 VP/cell. In polymerase chain reaction (PCR) experiments HC-AdV virus was used at a dose of 1000 VP/cell, wtAd5 virus at a dose of 10 VP/cell, and ΔE4 virus at doses from 1 to 200 VP/cell.
The methylase-expressing cell lines 293M and 116M were generated from HEK-293 and 116 cells (Palmer and Ng, 2003) transfected with plasmids encoding PaeR7 methyltransferase and a selection marker (hygromycin or puromycin), respectively. Stable 293M and 116M cell clones were grown for 3 weeks under selection pressure. Single stable cell colonies of 293M cells were further expanded in hygromycin (100 μg/ml)-containing selection medium and 116M cells in puromycin (750 ng/ml)-containing selection medium. 116M cells were routinely grown with DMEM supplemented with 10% FBS, hygromycin (100 μg/ml) for selection of Cre expression and puromycin (750 ng/ml) for stable PaeR7 methylase expression. 293M cells were grown in MEM supplemented with 10% FBS and hygromycin (100 μg/ml) for selection of PaeR7 methylase-expressing cells.
Methylated human wtAd5 was produced in 293M cells, and methylated HC-AdV was amplified in a suspension system (Palmer and Ng, 2003) using 116M cells.
Titration of adenoviral vector preparations
We determined the physical and infectious titers of all our adenoviral preparations (see also Fig. 1A). The physical titer equals the concentration of total genomes in a vector preparation and is expressed as viral particle (VP) numbers. It was routinely obtained by measuring the absorbance at 260 nm (OD260), which basically measures the total amount of viral DNA in a vector preparation. Viral DNA was released from virions obtained from CsCl gradients in Tris–EDTA (TE) buffer with 0.1% sodium dodecyl sulfate (SDS). The OD260 was measured to determine the viral titer according to the following formula: VP/ml = (absorbance at 260 nm) × (dilution factor) × (1.1 × 1012) × (36 kb)/(size of adenoviral genome in kb). Throughout this paper we refer to these VP numbers.

Characterization of high-capacity adenoviral vectors. (
Moreover, we directly compared infectious and physical adenoviral titers of wtAd, ΔE4 mutant virus, and HC-AdV in final vector preparations by quantitative real-time PCR (qPCR). This was important for direct comparison of results obtained with different viruses. For titration of wtAd, ΔE4 mutant virus, and HC-AdV we performed qPCRs that were run on a LightCycler system (Roche, Mannheim, Germany) using oligonucleotides Adfw1-24 (5′-CAT CAT CAA TAA TAT ACC TTA TTT-3′) and reverse436 (5′-ACG CCA CTT TGA CCC GGA ACG-3′) with LightCycler FastStart DNAMasterPLUS SYBR green I (Roche). The program was set as follows: preincubation at 95°C for 10 min and amplification in 45 cycles at 95°C for 10 sec, 55°C for 5 sec, and 72°C for 30 sec.
We also quantified helper virus contamination levels in HC-AdV preparations, using a PCR-based approach. Quantitative real-time PCR for helper virus contamination was carried out with the TaqMan 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA), amplifying an area of the adenoviral late gene 3 (L3) according to Puntel and colleagues (2006). Using oligonucleotides L3 forward (5′-AGA AGC TTA GCA TCC GTT ACT CGA GTT GG-3′; 400 nM) and L3 reverse (5′-ATA AGC TTG CAT GTT GGT ATG CAG GAT GG-3′; 400 nM) together with an L3-specific probe (5′-FAM-CCA CCC GTG TGT ACC TGG TGG ACA-TAMRA-3′; 300 nM) (Ella Biotech), the PCR was run with the following program amplifying a 235-bp fragment: AmpErase UNG reaction at 50°C for 2 min, preincubation/activation at 95°C for 10 min, and amplification and data collection in 40 cycles at 95°C for 15 sec and 60°C for 1 min. Universal PCR master mix (Roche) was used for TaqMan qPCR.
For determination of total particles, 10 VP/cell was directly administered to pelleted 293 cells before starting Hirt extraction. Subsequently, total particles were determined, analyzing 200 fg of Hirt-extracted DNA by qPCR. Infectious particles were determined by analyzing 200 fg of Hirt-extracted DNA of infected (10 VP/cell) 293 cells. These infected 293 cells were harvested with trypsin after 4 hr of incubation to remove unabsorbed virions.
Pulsed-field gel electrophoresis
For pulsed-field gel electrophoresis (PFGE) experiments 106 infected cells were removed from culture plates with trypsin. Pelleted cells were washed with Dulbecco's phosphate-buffered saline (GIBCO brand DPBS; Invitrogen) and resuspended in 0.25 ml of DPBS, which was immediately mixed with 0.25 ml of liquid 2% low melting point InCert agarose (Biozym Diagnostik, Cat. No. 850121, Oldendorf, Germany). This solution was cast into blocks and, after solidification, incubated at 55°C for 24 hr in 1% SDS and 125 mM EDTA with proteinase K at 5 mg/ml. The samples can be stored at 4°C in storage buffer (10 mM Tris, 10 mM EDTA). One-half of an agarose block was loaded on each line of the gel (Gold agarose; Biozym Diagnostik) and the pockets of the gel were sealed with InCert agarose (1%). All gels were electrophoresed at a ramped pulse frequency of 1–20 sec, at 6 V/cm2 for 24 hr (CHEF-DR III; Bio-Rad, Hercules, CA). Before Southern transfer, the gel was soaked in 0.2 M HCl for 30 min for depurination.
Southern transfer and hybridization
Hybond-N membranes (GE Healthcare Life Sciences, Piscataway, NJ) were used. Ultraviolet (UV) cross-linking was performed at 0.15 J/cm2. The membranes were hybridized in freshly prepared Church buffer (7% SDS, 0.5 M sodium phosphate [pH 7.5], 1 mM EDTA). For the first wash at 65°C we used washing buffer 1 (2% saline sodium citrate [SSC], 0.1% SDS) and for the second wash at 65°C we used washing buffer 2 (0.1% SSC, 0.1% SDS). The adenoviral ITR probe was a PCR product from bp 1 to 436 of the left arm of the adenoviral serotype 5 genome. The hFIX-specific probe (1.6 kb) was generated by cutting plasmid pAAV-EF1a-hFIX with HindIII and EcoRI (Nakai et al., 2000). Radioactivity from [α-32P]dCTPs was incorporated into the probes by random priming and Klenow DNA polymerase incubation (Stratagene, La Jolla, CA). Stripping of hybridized Southern membranes was performed with 0.4 M NaOH for 30 min at 42°C.
Hirt extraction of low molecular weight DNA
DNA was extracted from trypsinized cells. Tissue culture plates were washed once with cold DPBS, removed with trypsin, and pelleted. After that, cells were resuspended and lysed by adding 200 μl of lysis buffer (10 mM Tris, 10 mM EDTA [pH 7.5], 0.6% SDS). After an incubation period of 10 min at room temperature, 100 μl of 5 M NaCl was added and the plates were moved by gentle agitation for 2 min. The samples were incubated overnight at 4°C. After centrifugation at 13,000 rpm at 4°C for 40 min in a benchtop centrifuge, the pellets were discarded and 25 μg of proteinase K (34 U/mg of protein) was added to each supernatant and incubated at 37°C for 1 hr. Low molecular weight DNA was extracted twice by phenol–chloroform extraction. The viral DNA was precipitated at −80°C for at least 3 hr by adding 2 volumes of 100% ice-cold ethanol and 3 M sodium acetate (pH 5), followed by centrifugation at 13,000 rpm at 4°C for 1 hr. The pellets were washed with 70% ethanol and centrifuged for 5 min. DNA was resuspended in 100 μl of double-distilled H2O.
Isolation of genomic DNA from liver tissue
Two milliliters of lysis buffer (40 mM NaCl, 10 mM Tris [pH 7.5], 10 mM EDTA) was mixed with 300 μl of 10% SDS. Approximately 0.1 g of frozen liver tissue was cut with a scalpel and immediately transferred to the prepared lysis buffer. For homogenization, tissue in lysis buffer was pressed through a syringe several times. Afterward, 10 μl of proteinase K (20 mg/ml) was added and the samples were incubated at 55°C for 2 hr with permanent gentle shaking. Subsequently, 15 μl of RNase A (10 mg/ml) was added and the samples were incubated overnight at 37°C on a shaking platform. The next day, the samples were extracted twice with phenol–chloroform and after centrifugation at 3000 rpm for 2 min the supernatants were carefully transferred into fresh tubes. For precipitation of the DNA, 3 volumes of ice-cold 100% ethanol was added and the samples were mixed thoroughly. Subsequently, the samples were centrifuged at 2000 rpm for 10 min. Each supernatant was discarded and the DNA pellet was briefly air dried and resuspended in 500 μl of freshly prepared TE buffer (10 mM Tris [pH 7.5], 1 mM EDTA including RNase A [10 μg/ml]).
Polymerase chain reaction for concatemer and circular monomer detection
The HC-AdV concatemer/circle-specific PCR was run with 45 cycles at 95°C for 5 min, 95°C for 30 sec, 55°C for 30 sec, and 72°C for 2 min, using the oligonucleotides reverse436 (5′-ACG CCA CTT TGA CCC GGA ACG-3′) and Forward-1700 (5′-TCA GGC CAA GCT TAT CGA AAT TCC-3′). The ΔE4 mutant virus concatemer PCR was run under the same conditions with oligonucleotides reverse436 and H5dl1004For35585Circle (5′-CAC CAG CTC AAC AGT CAC AGT G-3′), but elongation was set at 72°C for 1 min. PCRs for control of infection were run under the same conditions, but with an elongation time of 30 sec, using oligonucleotides reverse436 and Ad5ITRfw1-24 (5′-CAT CAT CAA TAA TAT ACC TTA TTT-3′). If not stated otherwise, input of Hirt-extracted DNA was 400 ng in a total volume of 50 μl. To determine the sequence of PCR products, obtained PCR fragments were subcloned with the Zero Blunt TOPO PCR cloning kit (Invitrogen) and subsequently sequenced.
Analysis of HC-AdV replication by real-time PCR
XhoI cleavage was detected by running a PCR program with 40 cycles. The oligonucleotide 5′-FAM-TTC ACC GAG GGC CTA TTT CCC ATG AT-TAMRA-3′ was used as probe. The forward oligonucleotide was 5′-TCT GAG GCG GAA AGA ACC A-3′ and the reverse oligonucleotide was 5′-AAC AGC CTT GTA TCG TAT ATG CAA AT-3′. Probes and oligonucleotides were designed using Primer Express v3.0 software (Applied Biosystems).
Enzyme-linked immunosorbent assay
The human coagulation factor IX (hFIX) in murine serum was monitored by sandwich enzyme-linked immunosorbent assay (ELISA). A murine monoclonal IgG antibody (Sigma-Aldrich) directed against hFIX at a 1:2000 dilution was used for coating. As a secondary antibody we used the IgG antibody conjugated with horseradish peroxidase (Biozol Diagnostica Vertrieb, Eching, Germany) at a 1:1000 dilution. Peroxidase activity was detected at 492 nm, using SIGMAFAST OPD (phenylenediamine dihydrochloride) tablets (Sigma-Aldrich) and an ELISA reader (Tecan Group, Maennedorf, Switzerland).
Measurement of alanine aminotransferase
For quantification of alanine aminotransferase (ALT) levels in murine serum, we followed the instructions of the ALT (ALAT/GPT) detection kit (Randox Laboratories, Krumlin, UK). We applied 15 μl of murine serum to each reaction.
Animal studies
C57BL/6 mice were kept and treated according to the regulations of the government of Upper Bavaria in Germany. All mice were injected via the tail with a total volume of 200 μl. Virus was diluted in Dulbecco's phosphate-buffered saline (DPBS; Invitrogen). Rapid cell cycling of murine liver cells was induced by intraperitoneal administration of 50 μl of carbon tetrachloride (CCl4; Sigma-Aldrich) solution (1:1 dilution in mineral oil).
Results
Hepatic transfer of linear nonviral DNA and rAAV vectors was shown to result in long-term transgene expression. It is established that after transduction of target cells in vitro and in vivo rAAV and linear DNA form DNA concatemers by covalent linkage of vector genomes (Miao et al., 1998; Chen et al., 2003). These molecular structures contribute to the duration of vector DNA maintenance. Also, for HC-AdV-based gene transfer, multiple studies targeting the liver demonstrated long-term persistence of HC-AdV genomes and transgene expression. However, there is only limited information about molecular forms of adenoviral DNA molecules within a cell. HC-AdV genomes lack all viral coding sequences including E4. This feature of HC-AdV represents at least one prerequisite for concatemer formation, because it is established that on infection with recombinant adenovirus deleted for E4 ORF3 and ORF6, viral genomes are linked to one another at the termini (Weiden and Ginsberg, 1994).
Herein, we speculated that concatemerization or circle formation of HC-AdV DNA molecules and/or vector genome replication might contribute to the persistence of HC-AdV DNA and prolonged HC-AdV-derived transgene expression. Concatemeric HC-AdV DNA molecules could induce persistence because they may influence chromatin assembly of the DNA, or they may combine and multiply HC-AdV DNA-stabilizing effects. Improved stability of multimeric genomes may be mediated by combination and multiplication of cis-acting DNA-maintaining elements contained in the HC-AdV monomer (e.g., matrix attachment regions and centromeric regions, which are included in our HC-AdV genome).
Detailed characterization of adenoviruses used in this study
The adenovirus mutant H5dl1004 lacking parts of the E4 coding region and the HC-AdV FTC-hFIX-lucRNAi used in this study were described earlier (Sandler and Ketner, 1989; Rauschhuber et al., 2008). The genetic structures of all adenoviruses are summarized in Fig. 1A.
To allow direct comparison of our experiments, characterization of adenoviral vector preparations with respect to final titers was important. We first measured physical and infectious titers of all adenovirus preparations. We determined total genomes (physical titer expressed as viral particle units) by spectrophotometry and quantitative real-time PCR (qPCR). Furthermore, we measured and directly compared infectious genomes of wtAd5, ΔE4 mutant, and HC-AdV by qPCR (Fig. 1B). The ratio of infectious genomes to total genomes (viral particle units) was comparable for the HC-AdV and the ΔE4 mutant (black and gray bars in Fig. 1B). Although the physical titer measured by absorbance at 260 nm (OD260) was generally higher than the titer obtained by qPCR, the ratio of both parameters for all three viruses was similar (Fig. 1B). All viral doses applied in the present study relate to physical titer determinations by OD260 measurements expressed as viral particle (VP) numbers (for details, see Materials and Methods). Moreover, to achieve administration of comparable amounts of viral genomes, we directly compared and visualized the infectious ΔE4 mutant and HC-AdV titers by Southern blot analysis. We found that ΔE4 virus and HC-AdV vector show similar titers (Fig. 1C).
Because helper virus contamination in the HC-AdV preparation may potentially support HC-AdV replication in adenovirus early gene E1-complementing cell lines, it was also important to quantify helper virus contamination levels in final vector preparations. The contamination of HC-AdV preparations with helper virus was found to be at 0.09% as determined by qPCR (Fig. 1B).
Analysis of concatemer formation of high-capacity adenoviral vectors, using pulsed-field gel electrophoresis
To test whether concatemerization and/or circle formation of HC-AdV genomes lacking all viral coding sequences is possible and contributes to vector genome maintenance, we employed pulsed-field gel electrophoresis (PFGE) to resolve the high molecular weight viral DNA. In addition, we established a sensitive PCR approach. To define the detection limits of our PFGE system we performed dose-dependent studies with the ΔE4 virus H5dl1004 as a positive control for concatemer formation. We infected 293 cells and PFGE DNA profiles of viral genomes were analyzed. Southern blot analysis of the pulsed-field gel revealed at least dimeric concatemers starting at a dose of 6 VP/cell and strong concatemer formation was detected at high-dose (25 to 100 VP/cell) infection (data not shown). Monomeric genomes as well as dimeric and trimeric concatemers were detectable at 35, 70, and 105 kb.
To address whether HC-AdV forms concatemers, we infected 293 cells with the high-capacity adenoviral vector FTC-hFIX-lucRNAi (10,000 VP/cell). Control infections included the ΔE4 virus (H5dl1004) as a positive control for concatemer formation and wtAd5 virus as a negative control (100 VP/cell each). Infected 293 cells were harvested 24 hr postinfection and samples were subjected to PFGE analysis. After Southern transfer and hybridization of the blotted membranes with 5′ ITR-specific probe (detects HC-AdV, wtAd5, and ΔE4 virus) and hFIX-specific probe (detects only HC-AdV), exclusively monomers at the size of approximately 30 kb were detectable for HC-AdV genomes (Fig. 2, top). We detected a single band running at 36 kb for wild-type adenovirus and we observed concatemers for the ΔE4 virus. Notably, although we used a 100-fold higher dose for infection with HC-AdV the intensity of the band detected for HC-AdV was only slightly stronger compared with wtAd5 and ΔE4 virus, probably because of replication of wild-type and ΔE4 viruses in 293 cells. For detection of molecular structures of HC-AdV genomes at significantly lower dose the sensitivity of the PFGE assay was not high enough (data not shown). In summary, these results indicated that in contrast to ΔE4 mutant virus, high-capacity adenoviral vectors applied at high dose exist mainly as linear monomers in vitro. However, to analyze linkage of genomes by more sensitive means and at lower viral dose, a more sensitive assay was required.
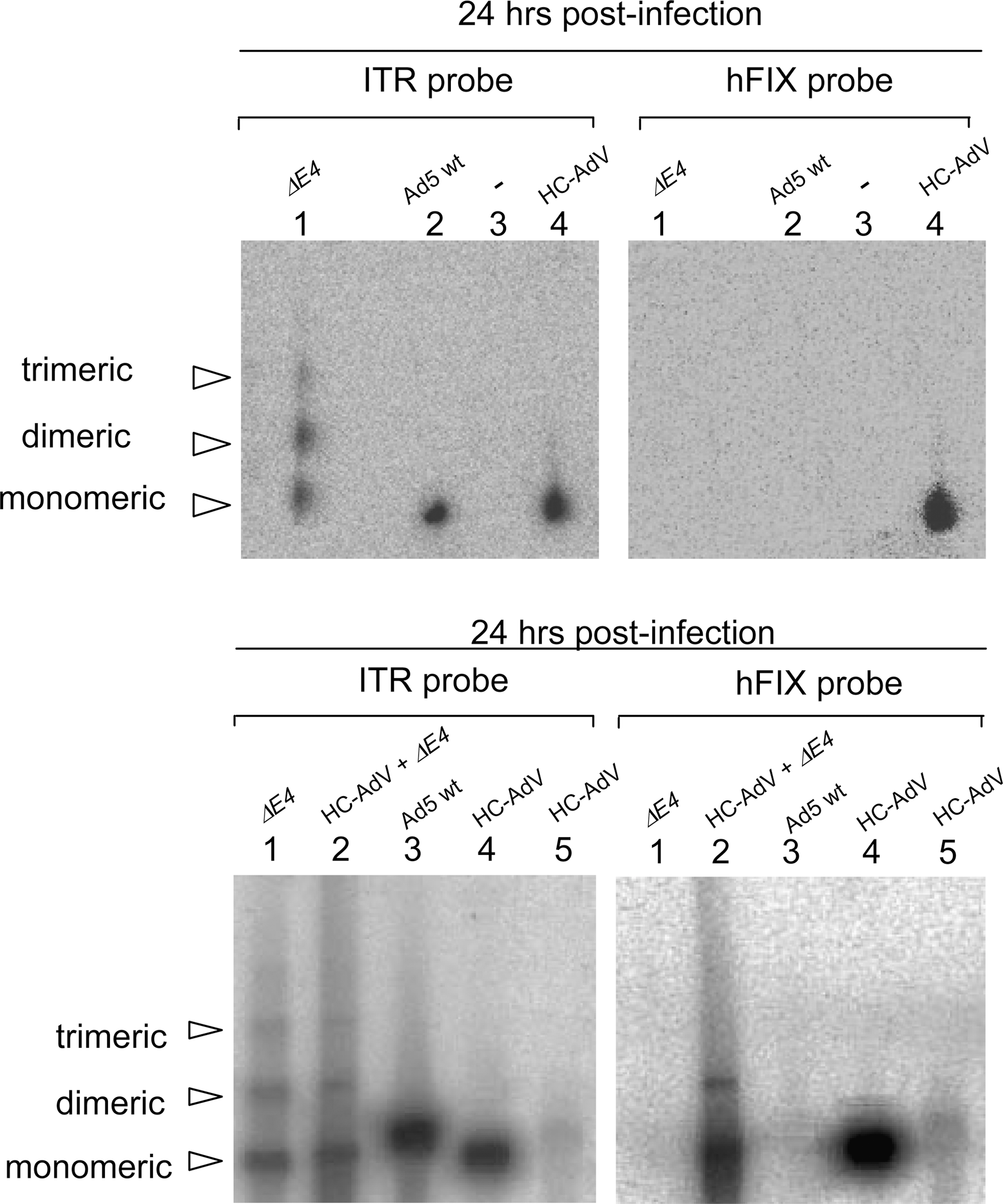
Molecular status of high-capacity adenoviral vector (HC-AdV) analyzed by pulsed-field gel electrophoresis (PFGE). Wild-type adenovirus (wtAd5), ΔE4 virus, and HC-AdV viral genomes were analyzed by PFGE and Southern blotting (top). Coinfection of HC-AdV and ΔE4 virus was performed to induce replication of HC-AdV and subsequent concatemerization (bottom). Cells were harvested 24 hr postinfection. The same Southern-blotted gel was hybridized with probes against the ITR (left, detects wtAd5, ΔE4 virus, and HC-AdV genomes) and hFIX (right, detects specifically HC-AdV). Top: Lane 1, 293 cells infected with ΔE4 virus at a dose of 100 VP/cell; lane 2, 293 cells infected with wtAd5 at a dose of 100 VP/cell; lane 3, genomic DNA of 293 cells; lane 4, 293 cells infected with HC-AdV at a dose of 10,000 VP/cell. Bottom: Lane 1, 293 cells infected with ΔE4 virus at a dose of 50 VP/cell; lane 2, 293 cells transduced with HC-AdV at a dose of 50 VP/cell and coinfected with ΔE4 virus at a dose of 50 VP/cell; lane 3, 293 cells infected with wtAd5 at a dose of 50 VP/cell; lane 4, 293 cells transduced with HC-AdV at a dose of 10,000 VP/cell; lane 5, 293 cells transduced with HC-AdV at a dose of 50 VP/cell.
As mentioned previously, the ΔE4 mutant virus showed strongest concatemer formation at high doses (data not shown). Because ΔE4 mutant virus replicates at high doses and shows low or no replication activity at low doses (Jayaram and Bridge, 2005), we hypothesized that replication may at least in part support linkage of HC-AdV adenoviral genomes. In addition, this may also provide a positive control for concatemer formation of HC-AdV when performing more sensitive assays. We coinfected 293 cells with HC-AdV and ΔE4 as helper virus at 50 VP/cell. In this experiment the ΔE4 virus H5dl1004 provides all the proteins necessary for replication of HC-AdV genomes in trans, but leaving out E4 keeps the DSBR machinery active. Southern-blotted PFGE gels were hybridized with probes against hFIX and ITR. In contrast to infection with HC-AdV alone, concatemeric HC-AdV genomes were detectable in the sample coinfected with ΔE4 virus (Fig. 2, bottom, lane 2). We concluded that coinfection with a ΔE4 virus and subsequent replication seems to facilitate concatemer formation of HC-AdV.
Detection of adenoviral concatemers, using a sensitive PCR system
To investigate the potential formation of concatemers by more sensitive means and to test the possibility of circularization of recombinant adenoviral vector genomes, we designed a PCR-based approach specific for concatenated and circular genome conformations. An outline of this PCR approach is schematically shown in Fig. 3A. Using this approach head-to-tail concatemers, head-to-tail circular monomers, and tail-to-tail or head-to-head concatemers can be discriminated. If neither concatemers nor circular monomers are present, no PCR product is generated.

Detection of concatemers and circular monomers by PCR. (
To define the detection limits of our PCR system we first performed dose-dependent studies with ΔE4 virus. We infected 293 cells with this virus at doses ranging from 1 to 200 VP/cell and extracted low molecular weight DNA by Hirt extraction. A smear of differently joined concatemers was detected starting from 3 VP/cell (Fig. 3B, top). Interestingly, at high viral dose (best visible at the dose of 200 VP/cell), in addition to the smear two bands appeared to be visible, indicating that predominant molecular forms of linkage of recombinant viral vector genome ends may exist. To demonstrate that all samples received the ΔE4 virus, we performed a second PCR with primers annealing at the 5′-ITR/ψ region of the adenoviral genome (Fig. 3B, bottom). To provide a sensitivity assay for our concatemer and control-of-infection PCR, we serially diluted the Hirt-extracted sample of 293 cells that was infected with ΔE4 virus at a dose of 3 VP/cell (see also Fig. 3B, top). The concatemer PCR was 10-fold less sensitive than the control-of-infection PCR and ΔE4 concatemers were detectable down to a 10−4 dilution (42 pg of DNA; data not shown). Furthermore, we determined the total genome copy number of ΔE4 genomes present in the respective samples. We found that 237 copies (in the 10−5 dilution) generate a detectable signal in the linear control-of-infection PCR; 2730 copies (in the 10−4 dilution) are needed to detect concatemers (data not shown).
To prove that the smear we obtained in the PCR truly consisted of differently joined ΔE4 concatemers, we subcloned the resulting DNA fragments appearing on the gel as a smear. Subsequently, we selected 15 clones, isolated plasmids, and sequenced the inserted ΔE4 DNA fragments. We found that every subclone showed a different sequence at its junctions, with large (up to 750 bp) or rather small deletions (down to 70 bp) in the ITR and/or ψ region. Representative junction examples are shown in Fig. 3C.
We next established a similar PCR specifically detecting concatemers and/or circles derived from HC-AdV genomes. Thus, low molecular weight DNA was isolated from 293 cells coinfected with HC-AdV and ΔE4 at 50 VP/cell. This experimental setup was chosen because, based on our PFGE experiments, we knew that this would result in HC-AdV genome concatemers (Fig. 2, bottom). A smear of differently joined HC-AdV genome concatemers was observed starting from a 10−4 dilution (42 pg of DNA; Fig. 3D, top). To demonstrate that all samples contain HC-AdV and ΔE4 virus, a second PCR was performed with primers annealing at the 3′ ITR region of the adenoviral genome and upstream of the 3′ end of the HC-AdV and ΔE4 genome, respectively (Fig. 3D, middle and bottom). To prove that the smear obtained by PCR truly consisted of differently joined HC-AdV concatemers, the resulting DNA fragments appearing on the gel as a smear were subcloned. Subsequently, we selected clones, isolated plasmids, and sequenced the inserted HC-AdV DNA. We found that every subclone showed a different sequence at its junction site, with large deletions (up to 2000 bp). Representative examples are shown in Fig. 3E.
Analysis of concatemer formation of HC-AdV in vitro
After establishment of the sensitive concatemer/circle-specific PCR for ΔE4 and HC-AdV virus, we addressed whether HC-AdV forms circles and/or concatemers. Furthermore, we investigated whether the molecular status may be cell line dependent. We infected 293 cells, HeLa cells, and various hepatic cell lines (SK-HEP-1, HuH-7, and murine Hepa 1A) with HC-AdV FTC-hFIX-lucRNAi at a dose of 1000 VP/cell. Low molecular weight DNA was isolated by Hirt extraction and the concatemer/circularization assay was performed. We found that in contrast to our positive controls no concatenated or circular genome conformations were visible 72 hr postinfection with HC-AdV (Fig. 3F, top). This finding was independent of the cell type used and the viral dose applied (data not shown). To demonstrate that all cell lines analyzed were actually transduced with the input virus, a control PCR was performed to detect linear monomers of viral vector DNA (Fig. 3F, bottom). We concluded that linear monomers are the predominant molecular form of HC-AdV in vitro. Thus, these results indicated that the infecting recombinant adenoviral DNA molecule was not subject to the DSBR machinery of the host cell in vitro.
Establishment of a methylase/restriction endonuclease-based system for analysis of HC-AdV replication
As a further mechanism of transgene persistence after liver-directed HC-AdV gene transfer we wanted to address whether replication may at least in part contribute to persistence of transgene expression. In our case the HC-AdV genome contains a centromeric region and a matrix attachment region, which may play a role in replication. Also, replication of HC-AdV genomes may be possible because of the unavoidable low-level helper virus contamination in final HC-AdV vector preparations, which may provide viral proteins required for replication in trans. In our case this helper virus contamination level is usually about 0.1%.
To test this hypothesis, we generated PaeR7 methylase-expressing 293 and 116 cells for the generation of methylated wtAd5 and first-generation adenoviral vectors (293M cells) and methylated HC-AdV genomes (116M cells). PaeR7 methylase adds a methyl group at the N6 position of the adenine base of XhoI sites (Gingeras and Brooks, 1983). In this system, viral replication restores XhoI cleavage by removal of methylation and replication can be detected by XhoI cleavage.
Both cell lines showed expression of PaeR7 methylase as demonstrated by reverse transcriptase PCR (data not shown). To test for methylase functionality, we produced wtAd5 and HC-AdVs in these cell lines. After CsCl purification, the virions were disintegrated and purified viral genomes were subjected to XhoI digest. Genomes were methylated as demonstrated by the absence of XhoI cleavage. However, the methylation had no effect on the ability to digest the DNA with EcoRI (Fig. 4A). The specific methylation of XhoI sites had no detectable influence on virus/vector titer yield and transgene expression (data not shown). To confirm that PaeR7 methylation is removed by replication, we infected 293 cells with methylated HC-AdVs at 1000 VP/cell. To promote replication of HC-AdV genomes we coinfected with helper virus at a dose of 10 VP/cell. Helper virus-driven replication of HC-AdV genomes was confirmed by Southern blotting, showing that PaeR7 methylation can be removed by de novo genome synthesis (Fig. 4B). Thus, our system can be used to monitor the replicative status of HC-AdV DNA molecules.

Replicative status of high-capacity adenoviral vector genomes (HC-AdV) in various cell lines. (
Replicative state of HC-AdV genomes in vitro
To analyze the replicative status of HC-AdV genomes in vitro, we infected various cell lines (293, HuH-7, SK-HEP-1, and Hepa 1A) with methylated HC-AdVs at doses ranging from 100 to 3000 VP/cell. Transduced cells were harvested 48 to 96 hr postinfection and low molecular weight DNA was analyzed by XhoI digest and subsequent Southern blotting. The HC-AdVs remained methylated and, therefore, did not replicate in any cell line tested (Fig. 4C). This result was independent of the viral dose applied. Moreover, this experiment implies that low-level helper virus contamination may not be sufficient to support HC-AdV replication in the cell types analyzed.
Molecular status of HC-AdV vector genomes in quiescent and cycling cells in vivo
In several liver-based gene transfer studies using different animal species it was demonstrated that after a single injection of HC-AdV long-term persistence of viral DNA molecules and transgene expression can be achieved (Morral et al., 1998, 1999; Schiedner et al., 1998; Kim et al., 2001; Ehrhardt and Kay, 2002; Ehrhardt et al., 2003a; Toietta et al., 2005; Brunetti-Pierri et al., 2007). To address whether the vector DNA structure is involved in persistence, we aimed at analyzing the molecular status of HC-AdV DNA molecules in murine liver. We have chosen C57BL/6 mice because we and others showed long-term transgene expression and vector genome persistence in this mouse strain (Belalcazar et al., 2003; Ehrhardt et al., 2007).
To investigate whether concatemers and/or circular monomers are formed in vivo and whether HC-AdV genomes show any replication activity in murine liver, we injected 7.5 × 109 VP of methylated HC-AdV into the tail vein of C57BL/6 mice (n = 8). In addition to the molecular status of viral genomes in quiescent/slowly proliferating hepatic tissue, we wanted to determine whether the induction of liver cell proliferation influences the molecular forms and the replicative state of HC-AdV DNA. It was demonstrated that the expression profile of hepatocytes significantly changes during induction of mitosis (Fukuhara et al., 2003). This may indirectly lead to conformational changes of adenoviral DNA molecules or might promote HC-AdV replication. Therefore, we intraperitoneally administered carbon tetrachloride (CCl4) 6 weeks after viral infection (n = 3). CCl4 leads to liver necrosis (Weber et al., 2003). To reconstitute normal liver size and function the treated liver undergoes cell cycling and regenerates. These CCl4-treated mice were killed 7 weeks (mouse 1) or 9 weeks (mice 4 and 5) postinjection. Another group of mice received HC-AdV alone (n = 5) and murine livers were isolated 5 weeks (mice 2 and 3) and 9 weeks (mice 6, 7, and 8) postinjection. As a negative control group two mice (no. 9 and 10) were mock infected with Dulbecco's phosphate-buffered saline (DPBS) containing no viral particles.
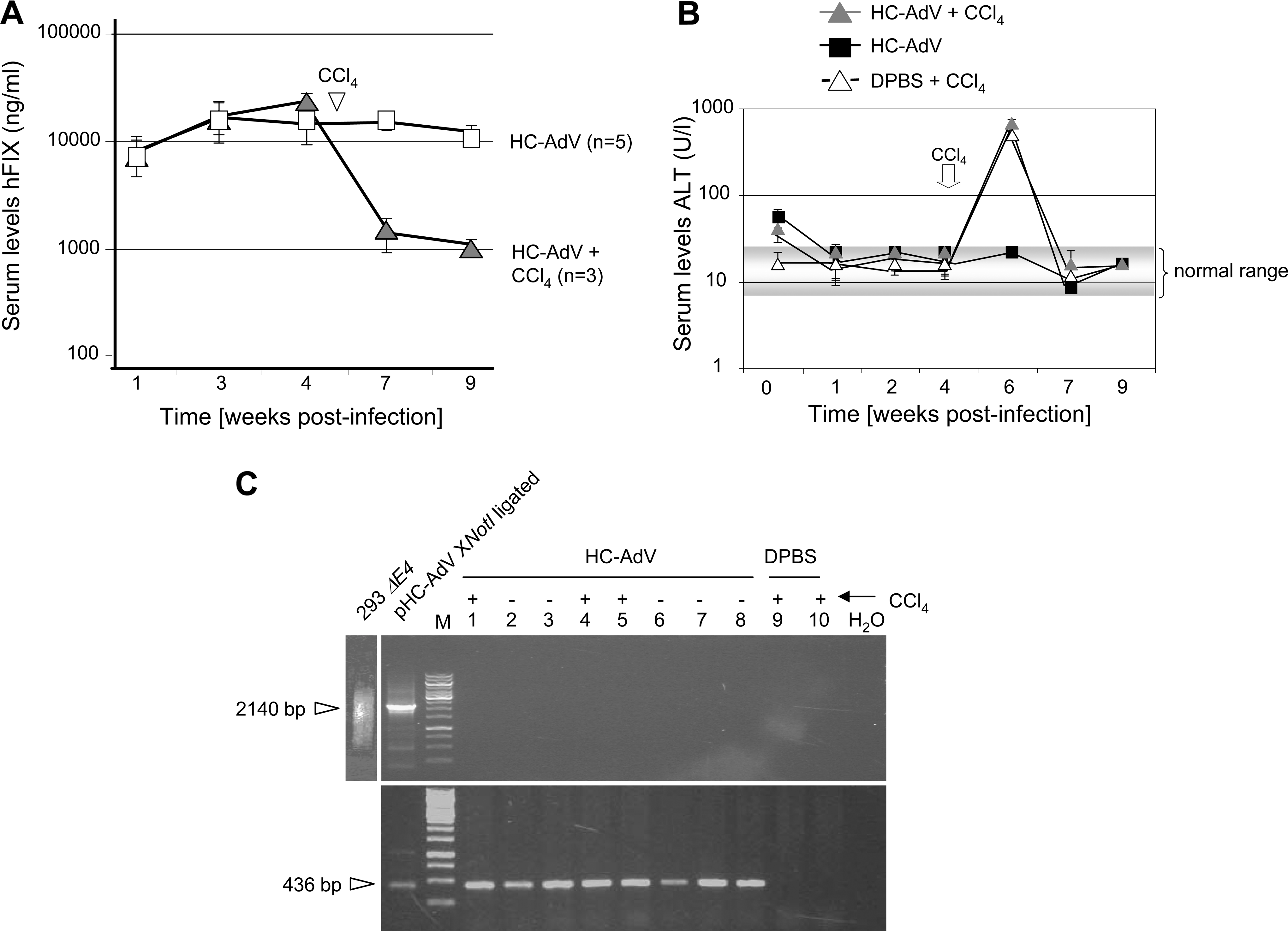
To survey the course of the experiment, all three groups were monitored for serum levels of the human blood coagulation factor IX (hFIX) expressed from HC-AdV (Fig. 5A). We also analyzed alanine aminotransferase (ALT) levels (Ozer et al., 2008) as an indicator of liver toxicity for up to 9 weeks postinfection. After an initial peak of ALT levels on the first day postinfection, liver enzyme levels stayed within the normal range for all non-CCl4-treated animals. In contrast, CCl4-treated mice showed high levels of alanine aminotransferase after drug administration, which declined to normal levels shortly thereafter (Fig. 5B). After injection of CCl4 we also observed a rapid decline in serum hFIX levels, indicating that extensive liver cell cycling occurred.
Characterization of high-capacity adenoviral vector (HC-AdV) DNA molecules in liver of C57BL/6 mice. C57BL/6 mice were infected with 7.5 × 109 transducing units of the high-capacity adenoviral vector FTC-hFIX-lucRNAi. To induce rapid cell division in mouse liver carbon tetrachloride (CCl4) was injected. The uninfected control group received Dulbecco's phosphate-buffered saline (DPBS). (
Mice were killed and murine livers were collected to isolate genomic liver DNA, including HC-AdV DNA. To analyze concatemer and circle formation of HC-AdV, this purified DNA was subjected to a PCR protocol as described previously (Fig. 3A). In concordance with our in vitro results, we found that the PCR did not visualize specific products of concatenated or circular vector genomes in any of the HC-AdV-infected animals (Fig. 5C, top). All mice contained HC-AdV genomes as shown by PCR (Fig. 5C, bottom). No PCR product was obtained in the negative control group (mice 9 and 10). Thus, our results indicated that linear monomers are the predominant molecular form of HC-AdV genomes in murine liver.
To analyze the replicative status of HC-AdV in murine liver and to detect even low amounts of viral DNA that might have lost methylation by replication, we established a sensitive real-time PCR assay with oligonucleotides that anneal around a specific XhoI site within the HC-AdV genome. As a control experiment, we investigated replication kinetics to confirm that our methylase/endonuclease-based system is applicable to our real-time PCR assay. To this end, 293 cells were transduced with methylated HC-AdV at a dose of 100 VP/cell and with helper virus at a dose of 10 VP/cell to promote replication of HC-AdVs. We harvested cotransduced cells at the time points indicated, isolated low molecular weight DNA, and digested the samples with XhoI or NcoI. NcoI cleaves HC-AdV DNA at arbitrary positions but not between real-time PCR primer annealing sites. At 1 and 6 hr postinfection the methylated input DNA was reisolated and no difference between XhoI and NcoI digest was detectable (Fig. 6A). At 12 hr postinfection, replication and demethylation of HC-AdV had started, indicated by a shift of the crossing point of the XhoI-digested sample to higher PCR cycle numbers. Twenty-four and 48 hr postinfection, methylated input HC-AdV DNA was no longer detectable, demonstrating that at these time points replication promoted by helper virus proteins occurred (Fig. 6A). Notably, the total amount of HC-AdV DNA within these samples was increased, indicated by a shift of the crossing points of the NcoI-digested samples to lower PCR cycle numbers. As additional controls, we ran digested unmethylated plasmid DNA (pAdFTC) and CsCl-purified methylated HC-AdV DNA.

HC-AdV vectors have no replication activity in murine liver. (
To analyze the replicative status of HC-AdV in treated C57BL/6 mice (Fig. 5A and B), we digested genomic liver DNA with XhoI and, as a control, with equal amounts of NcoI as described previously. In both animal groups treated with HC-AdV plus CCl4 or HC-AdV alone, no difference in XhoI-digested and NcoI-digested samples was detectable (Fig. 6B). Thus, the methylated input HC-AdV genomes do not undergo replication in quiescent and mitotic hepatocytes. In summary, persistence of HC-AdV genomes is independent of DNA replication.
Discussion
The rationale for our study was to unravel a possible mechanism for the persistence of HC-AdVs. We and others (Schiedner et al., 1998; Ehrhardt and Kay, 2002; Kreppel et al., 2002; Kreppel and Kochanek, 2004; Jozkowicz and Dulak, 2005; Brunetti-Pierri et al., 2007) showed that after a single injection of HC-AdV particles, transgene expression and the therapeutic effect are maintained long term and thus also the viral genome. One study even showed sustained transgene expression in nonhuman primates for up to 964 days, using a minimally invasive method based on a balloon occlusion catheter (Brunetti-Pierri et al., 2009). Even after induction of cell cycling of the host cell in murine liver the HC-AdV DNA molecule seems to be more stable compared with nonviral DNA (Ehrhardt et al., 2003b).
We speculated that several mechanisms or molecular forms of HC-AdV DNA molecules may potentially contribute to the persistence of HC-AdV genomes. The present studies provide some support that replication and concatemer or circle formation can be ruled out as potential mechanisms participating in vector genome maintenance.
An infection with adenovirus exposes the host cell to exogenous linear double-stranded DNA (dsDNA) and leads to the activation of cellular DSBR proteins (Boyer et al., 1999; Stracker et al., 2002; Weitzman, 2005). The Mre11–Rad50–Nbs1 (MRN) complex is involved in both, homologous recombination (HR) and nonhomologous end joining (NHEJ), whereas the DNA-dependent protein kinase (DNA-PK) is the key constituent involved in NHEJ. The adenoviral proteins E4 34 kDa and E4 11 kDa prevent the DSBR system from “repairing” the viral genome by physically interacting with DNA-PK (Boyer et al., 1999). In addition, the E4 34-kDa protein forms a complex with a viral 55-kDa protein encoded by the adenoviral early region 1b. This complex targets the DSBR proteins of the Mre11–Rad50–Nbs1 complex, forming a substrate for proteasome-mediated degradation (Stracker et al., 2002). Therefore, the genomes of ΔE4 mutants are connected during the viral replication process, leading to multimeric chains of adenoviral DNA. Our hypothesis, that HC-AdV genomes are also affected by the DSBR system, could not be confirmed because we were not able to detect concatemers. This finding raises questions concerning whether other proteins that are provided by ΔE4 mutants but not by HC-AdV contribute to concatemer formation. Candidate proteins—both viral (pTP, Ad-Pol, and DBP) and cellular (Oct1 and NFI) (de Jong and van der Vliet, 1999)—that interact at the ITRs of the adenoviral genome could enhance the recruitment of DSBR proteins to the viral termini.
Interestingly, the junctions between the ΔE4 virus and replicative HC-AdV genomes are diverse (Fig. 3). This phenomenon cannot be explained by HR or simple ligation of vector genome termini. As it is known that NHEJ leads to random deletions at the DNA junctions (Allen et al., 2003), it is likely that NHEJ may represent the predominant mechanism of ΔE4 and HC-AdV concatemer formation.
Results presented in this study are in contrast to other gene transfer systems based on linear DNA or recombinant AAV. With these vector systems linear DNA fragments are introduced into the cell, providing a substrate for the DSBR system, which leads to linkage of free DNA vector ends. In that respect HC-AdV, which also enters the cell as a linear DNA monomer, seems to be unique because it persists as a linear monomer. We speculate that the terminal protein (TP), which is covalently bound to the ITR, or cellular proteins interacting with the ITR may prevent concatemer formation.
We found that coinfection with a ΔE4 mutant virus facilitates concatemer formation of HC-AdV genomes (Fig. 2, bottom and Fig. 3D). This raises questions concerning whether replication may represent a prerequisite for linkage of HC-AdV genomes, because coinfection of HC-AdV with ΔE4 mutant virus most likely supports replication of HC-AdV. The same phenomenon may also hold true for ΔE4 mutant virus. At a high dose, ΔE4 adenoviral mutants are able to initiate amplification of viral DNA because of expression of E1 and E2 proteins and cellular factors. This leads to accumulating amounts of viral DNA inside the host cell that is subsequently concatenated (Sandler and Ketner, 1989; Jayaram and Bridge, 2005). At a low dose ΔE4 mutant virus is replication deficient and we observed that under these conditions linkage of genomes is less pronounced (data not shown). Notably, we also detected no concatemer formation for HC-AdV in 293 and HeLa cells. In theory, concatemer formation in these cell types may have been possible, because in concert with low-level helper virus contamination and a cell line providing the function of the adenoviral early gene E1 (293 cells and HeLa cells), replication of HC-AdV DNA molecules may occur. However, we also found that even high-dose low-level helper virus contamination may not be sufficient to support HC-AdV genome replication and subsequent concatemer formation (Fig. 2, top and Fig. 4C). Another reason for the fact that neither concatemer nor circle formation was observed may involve expression of the early gene E4 from the helper viral genome. This may have been sufficient to block formation of HC-AdV genome concatemers.
Moreover, we investigated the possibility of E1/E2-independent HC-AdV replication as a mechanism of persistence. Because our HC-AdV contains a putative mammalian origin of replication originated from a centromeric region (Ehrhardt and Kay, 2002), our hypothesis was that the mammalian origin of replication could be activated by replication of the host cell machinery. However, in replication-activated hepatocytes that were treated with CCl4, and in quiescent liver tissue, no replication of HC-AdV genomes was observable. Notably, the small percentage of contaminating ΔE1/E3 helper virus that is copurified during vector preparation did not initiate replication of HC-AdV in murine liver either. These results are in concordance with the findings from Nelson and Kay (1997). They reported replication of first-generation vectors in vitro but not in vivo. Remarkably, the nonreplicative state of the HC-AdV genome despite the helper virus contamination may make the use of HC-AdV in gene therapy even safer.
Our results differ from those of Kreppel and Kochanek (2004), who found circular molecules of their HC-AdV vector. They discovered spontaneous circularization of the HC-AdV in the absence of Flp-mediated circularization. The vector encoding EBNA1/oriP showed the best results in this respect. This suggests that also DNA sequences contained in the vector might have yet unknown significant features that contribute to circularization. Moreover, Ruben and colleagues (1983) provided evidence that adenovirus serotype 5 DNA molecules may at least in part form covalently closed circles. However, it was speculated that concatemers and/or circular forms of adenovirus serotype 5 genomes in primary rat kidney cells may represent intermediates during the integration process.
We speculated that several mechanisms or molecular forms of HC-AdV DNA molecules may potentially contribute to the persistence of HC-AdV genomes (Ehrhardt et al., 2003b, 2008; Jager and Ehrhardt, 2007). The HC-AdV genome could (1) replicate episomally, (2) possess a nuclear retention signal or a centromeric region that results in an improved association with chromatin, (3) form concatemers or (4) circularize, (5) integrate, or (6) associate with histones. We were able to rule out replication and concatemer/circle formation. Future studies need to investigate other possibilities of HC-AdV genome maintenance.
Taken together, we have demonstrated that, in contrast to AAV vectors and linear extrachromosomal DNA, HC-AdV exists predominantly as linear monomers. Furthermore, we have shown that the originally transduced HC-AdV genomes persist without replication in vitro and in vivo. These findings add to the already improved safety profile of HC-AdV compared with earlier vector generations.
Footnotes
Acknowledgments
H5dl1004 ΔE4 mutant virus was provided by Gary Ketner (Johns Hopkins University, Baltimore, MD). The ΔE4-producer cell line W162, originally also from Gary Ketner, was kindly provided by Thomas Dobner (Heinrich Pette Institute, Hamburg, Germany). The authors are grateful to Ulrich Koszinowski, Christina Rauschhuber, and Nicola Wolf for critical reading of the manuscript. Furthermore, the authors thank Philip Ng for providing the producer cell line 116 and the helper virus AdNG163R-2 for high-capacity adenoviral production. This work was supported by DFG grant SFB455 to A.E.
Author Disclosure Statement
No competing financial interests exist for any of the authors of this paper.