
Abstract
Strategies to improve vaccine efficacy are still required, especially in the case of chronic infections, including human immunodeficiency virus (HIV). DNA vaccines have potential advantages over conventional vaccines; however, low immunological efficacy has been demonstrated in many experiments involving large animals and in clinical trials. To improve the immunogenicity of DNA vaccines, we have designed a plasmid vector exploiting the binding capacity of the bovine papillomavirus E2 protein and we have used electroporation (EP) to increase DNA uptake after intradermal inoculation. We demonstrated, in nonhuman primates (NHPs), efficient induction of anti-HIV immunity with an improved DNA vaccine vector encoding an artificial fusion protein, consisting of several proteins and selected epitopes from HIV-1. We show that a DNA vaccine delivery method combining intradermal injection and noninvasive EP dramatically increased expression of the vaccine antigen selectively in the epidermis, and our observations strongly suggest the involvement of Langerhans cells in the strength and quality of the anti-HIV immune response. Although the humoral responses to the vaccine were transient, the cellular responses were exceptionally robust and persisted, at high levels, more than 2 years after the last vaccine boost. The immune responses were characterized by the induction of significant proportions of T cells producing both interferon-γ and interleukin-2 cytokines, in both subpopulations, CD4+ and CD8+. This strategy is an attractive approach for vaccination in humans because of its high efficacy and the possible use of newly developed devices for EP.
Introduction
Vaccination is one of the most attractive strategies to prevent infections and to control epidemics. However, there are still no effective vaccines against many infectious diseases, including emerging and chronic infections (Plotkin, 2005). It is thought that a successful strategy for halting the human immunodeficiency virus (HIV) pandemic will require an effective prophylactic vaccine that induces as broad an immune response as possible. Attention has focused on immunogens that can generate long-term memory CD8+ T cells recognizing conserved viral epitopes. This strategy potentially arms the host with immunity that controls viremia and disease. Despite the failure in a phase 2b study of a T cell-based vaccine (Cohen, 2007; Buchbinder et al., 2008; Sekaly, 2008), there is evidence that CD8+ T cell responses play a key role in containing early HIV replication (Borrow et al., 1994, 1997; Koup et al., 1994; Klein et al., 1995; Ogg et al., 1998; Rowland-Jones et al., 1998) and can control disease progression (Ogg et al., 1999; Wagner et al., 1999). More recently, immune control of a simian immunodeficiency virus (SIV) challenge has been achieved in rhesus macaques with a T cell-based vaccine, using a combination of adenoviral vectors (Liu et al., 2009). Strategies for inducing cellular immune responses may therefore be required. Preexisting vector-specific neutralizing antibodies have been reported to reduce the immunogenicity of live attenuated vector-based vaccines (Lin et al., 2008; Priddy et al., 2008) and may compromise their safety (Cohen, 2007). Genetic vaccines such as plasmid DNA are potentially valuable tools that could overcome theses inconveniences.
Nonhuman primates (NHPs) are particularly valuable for assessing human vaccines and have been used to study many recombinant vectors including plasmid DNA vaccines (Boyer et al., 1996; Lu et al., 1996; Lekutis et al., 1997; Barouch et al., 2000; Couillin et al., 2001), and live attenuated vectors such as the modified vaccinia virus strain Ankara (Amara et al., 2001), NYVAC (Benson et al., 1998), adenovirus (Shiver et al., 2002; Patterson et al., 2003), and Semliki Forest virus (Mossman et al., 1996; Berglund et al., 1997; Martinon et al., 2008). Many other vectors have been constructed (reviewed in Girard et al., 2006) and live attenuated vectors have generally successfully induced immune responses predominantly mediated by T lymphocytes. However, these responses were directed not only against the recombinant antigens, but also against the vector itself and preexisting T cell-mediated immunity to the vector has been shown to compromise vaccination efficiency (Sharpe et al., 2001).
DNA-based vaccines represent an attractive alternative to other modes of vaccination. The in vivo synthesis of DNA vaccine-encoded proteins mimics expression of antigens after viral infection and may improve processing and presentation to the immune system, thereby providing the advantages of live attenuated vaccines without the safety and stability concerns associated with the administration of infectious agents. Moreover, they are easy to produce and offer many possibilities for large inserts and expressing systems. However, despite promising results reported in small laboratory animals, low immunological efficacy has been demonstrated in large animal models and appeared to be lower than desired in human clinical trials (reviewed in Liu et al., 2006; Lu et al., 2008). However, DNA vaccine immunogenicity is strongly improved by the electroporation (EP) treatment of the injection site in NHPs such as macaques (Glasspool-Malone et al., 2000; Otten et al., 2004; Luckay et al., 2007; Cristillo et al., 2008; Hirao et al., 2008a,b; Laddy et al., 2008; Rosati et al., 2008) and in pigs (Glasspool-Malone et al., 2000; Babiuk et al., 2003, 2004; Hirao et al., 2008b).
In this study we have used a novel DNA vector, auxo-GTU-MultiHIV-B (Blazevic et al., 2006). We show here that EP treatment of the vaccine injection site dramatically increases expression of the vector-encoded antigen in the skin and subsequent immune responses in macaques.
Materials and Methods
Construction and purification of auxo-GTU-MultiHIV-B DNA vaccine
The prototype HIV vaccine we developed is based on an auxo-GTU vector with a tailor-made MultiHIV-B antigen insert encoding clade B HIV sequences derived from Rev, Nef, Tat, p17, and p24 proteins and a stretch of tandemly arranged T cell epitope-rich clusters from the Pol and Env proteins (Fig. 1).The molecular mass of the produced fusion protein was about 120 kDa, which corresponds to the 1064 amino acids encoded by the construct (Blazevic et al., 2006). The backbone variant of GTU, auxo-GTU, enables production by antibiotic-free, stable, and large-scale fed-batch fermentation. Escherichia coli strain AG1 ΔaraD carrying the vaccine vector was grown by fed-batch culture in the presence of l-arabinose. Bacterial lysate was prepared by an alkaline method according to standard procedures. auxo-GTU-MultiHIV-B plasmid and auxo-GTU-empty plasmid DNAs were prepared by three-step chromatography purification (GE Healthcare Life Sciences, Piscataway, NJ). The vaccine was diluted in phosphate-buffered saline (PBS) to a final concentration of 1 mg/ml for the final vaccine formulation. The quantitative expression of marker genes was measured after transfection of Jurkat cells or concanavalin A (ConA) blasts, which were obtained from macaque peripheral blood mononuclear cells (PBMCs) freshly isolated and cultured in RPMI 1640 medium with GlutaMAX I (Invitrogen, Cergy Pontoise, France) supplemented with 10% heat-inactivated fetal calf serum (FCS; Laboratoires Eurobio, Les Ulis, France) (culture medium) and ConA at 5 μg/ml (Sigma-Aldrich, Saint-Quentin Fallavier, France) for 2 days before transfection. Primary lymphoblast cell lines were then adjusted to 106 cells/ml in culture medium supplemented with interleukin (IL)-2 at 20 IU/ml (Roche Diagnostics, Meylan, France).
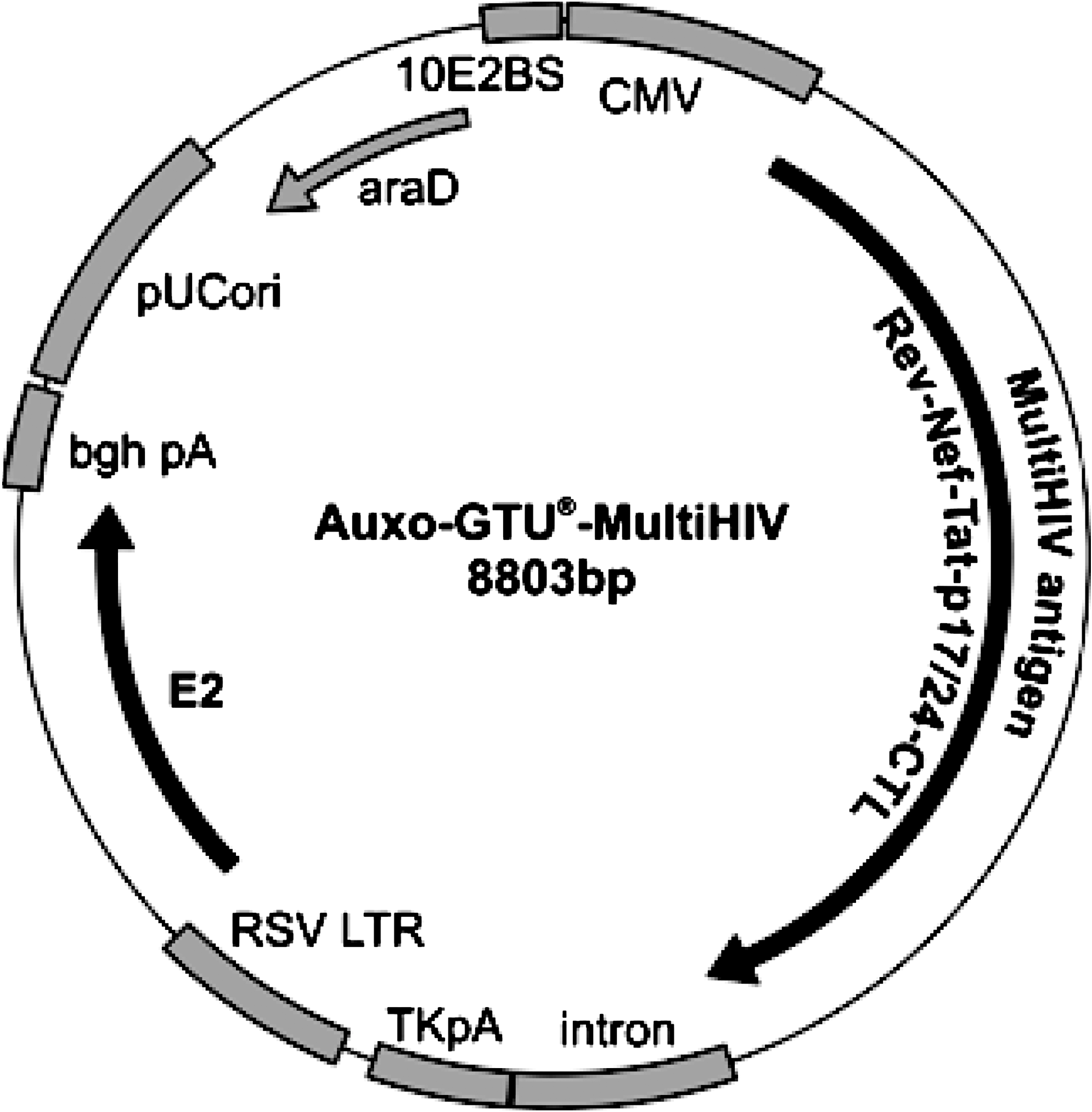
Map of the auxo-GTU-MultiHIV plasmid. The vector consists of two expression cassettes, the first encoding the antigenic fusion protein called MultiHIV (driven by the cytomegalovirus [CMV] promoter) and the second encoding the auxiliary protein E2 from bovine papillomavirus (driven by the Rous sarcoma virus [RSV] promoter). MultiHIV is an artificial fusion protein, having a total length of 1064 amino acids and representing full-length sequences of Rev, Nef, Tat, p17, and p24 (located at the N-terminal part of the MultiHIV antigen). Selected T cell epitopes from protease, reverse transcriptase, and envelope are located at the C-terminal part of the antigenic protein, designated as CTL. The vector does not carry any antibiotic resistance gene, and the araD gene provides positive selection for plasmid-carrying bacteria in media containing l-arabinose. bgh pA, bovine growth hormone polyadenylation signal; pUCori, pUC origin of replication; RSV LTR, Rous sarcoma virus long terminal repeat; TKpA, HSV thymidine kinase polyadenylation sequence.
Animals and vaccinations
Twelve adult male cynomolgus macaques (Macaca fascicularis) from Mauritius, weighing 3 to 6 kg, were maintained and handled in accordance with European guidelines for NHP care (EEC Directive N 86–609, November 24, 1986). The animals were sedated with ketamine chlorhydrate (10–15 mg/kg) for immunizations and collection of blood samples. Four macaques were immunized with auxo-GTU-MultiHIV-B plasmid by intradermal injection: each animal received 1 mg per immunization divided over 10 injection sites (100 μl per site). Immunizations were administered on weeks 0, 4, and 12, resulting in a cumulative dose of 3 mg per animal. Four macaques were immunized by intradermal injection followed by EP of the injection sites. The injection sites in this case were selected such that pairs of closely spaced sites could be folded between tweezer electrodes and electroporated together. As a result, these animals received injections at 10 sites and 5 EPs. EP consisted of six successive 10-msec square-wave pulses, output current 300–600 mA, with 90-msec intervals between pulses. These parameters were tested previously in a small pilot experiment and found to cause no tissue damage and to increase uptake of reporter plasmid into skin cells. A portable pulse generator (CUY21 EDIT; Nepa Gene, Ichikawa, Chiba, Japan) and tweezer electrodes were used, with a conductive gel to ensure good contact between the electrodes and skin. Four additional macaques received the same vector but without antigenic insert (empty vector): two of these control macaques received the vector as a simple intradermal injection, and two animals also received EP as described previously.
Immunohistochemistry
Five-micrometer frozen skin sections were prepared with a cryostat at −30°C and stored at −80°C. Sections were thawed, dried, fixed in acetone for 5 min at room temperature, and washed with PBS (Laboratoires Eurobio). They were incubated for 30 min at room temperature in a PBS solution containing 5% goat serum (Dako, Trappes, France) for blocking. The sections were then incubated at a final concentration of 10 μg/ml, in PBS buffer containing 5% goat serum and 0.1% saponin, at room temperature for 1 hr, with the following mouse monoclonal antibodies: anti-CD1a and anti-HLA-DP, DQ, DR (Dako), labeled with a Zenon mouse IgG1 Alexa Fluor 594 labeling kit (Molecular Probes/Invitrogen); anti-p24 (kind gift from B. Verrier, Institut de Biologie et Chimie des Protéines, UMR 5086 CNRS/UCBL, Lyon, France), labeled with a Zenon mouse IgG1 Alexa Fluor 488 labeling kit (Molecular Probes/Invitrogen); and anti-transglutaminase-1 (TGM1; LifeSpan Biosciences, Seattle, WA), labeled with a Zenon Alexa Fluor 647 labeling kit (Molecular Probes/Invitrogen). Negative controls for immunofluorescence staining involved replacing the primary antibody with isotype control, mouse IgG1 (BD Biosciences, Rungis, France). After intensive washing in 0.1% saponin in PBS and then in PBS, the skin sections were fixed in 4% formaldehyde, at room temperature for 15 min. The sections were washed in PBS, mounted with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories; CliniSciences, Montrouge, France), and stored in the dark at 4°C. Image analyses were performed with MetaMorph version 7.5 software (Molecular Devices, Sunnyvale, CA).
Antigen-specific T cells detected by enzyme-linked immunospot assay
MultiScreen 96-well filtration plates (Millipore, Guyancourt, France) were wet with 35% ethanol in water for 30 sec and then washed three times with sterile apyrogenic water and once with PBS. The plates were then coated by incubation overnight with monoclonal antibody against monkey interferon (IFN)-γ (clone GZ-4, 10 μg/ml; Mabtech, Sophia Antipolis, France) in PBS, at 4°C. Plates were washed six times with PBS and then blocked by incubation for 2 hr at 37°C in RPMI 1640 medium with GlutaMAX I (Invitrogen) supplemented with 10% heat-inactivated FCS (Laboratoires Eurobio) (culture medium). PBMCs were recovered by density gradient centrifugation and 2 × 105 cells were added to each well. Fifteen-mer overlapping peptides (11-amino acid overlap), tailored according to the sequence of the MultiHIV-B protein, were then added in triplicate to a final concentration of each peptide in the culture medium of 2 μg/ml. The peptides were subdivided into five different subpools, covering the sequence of Rev (29 peptides), Nef (51 peptides), Tat (25 peptides), and Gag p17/24 (91 peptides). Phorbol 12-myristate 13-acetate (Sigma-Aldrich) and ionomycin (Sigma-Aldrich), at final concentrations of 0.1 and 1 μM, respectively, were used as a positive control. Culture medium alone was used as a negative control. Plates were incubated for 18 hr at 37°C in a humid atmosphere containing 5% CO2. They were then washed five times with PBS and incubated overnight at 4°C with a 1-μg/ml concentration of biotinylated anti-IFN-γ antibody (clone 7-B6-1; Mabtech) in PBS containing 0.5% FCS. Plates were washed five times with PBS, incubated with alkaline phosphatase–streptavidin conjugate (0.25 μg/ml; Sigma-Aldrich) for 1 hr at 37°C, and washed five times with PBS. Spots were developed by adding 80 μl of NBT/BCIP substrate (Sigma-Aldrich) to each well. The spots were counted with an automated enzyme-linked immunospot (ELISpot) reader system with KS software (Carl Zeiss, Le Pecq, France). Results are expressed as means of IFN-γ spot-forming cells (SFCs) per 106 PBMCs (IFN-γ SFCs per million PBMCs) in triplicate wells. Background was calculated as the mean number of IFN-γ SFCs per million PBMCs in nonstimulated samples. Samples yielding more than 50 IFN-γ SFCs per million PBMCs after removal of the background were scored as positive.
Mapping of T cell epitopes, using individual peptides
T cell epitopes were mapped with cells of selected animals by ELISpot assays. The peptides were added to cells either individually (e.g., 28 different peptides covering Rev peptides), or arranged into several subpools according to matrix setup (91 peptides covering p17/24 protein were arranged into 19 subpools). Reacting peptides identified with subpools were confirmed in individual assays.
Depletion of T cell subpopulations from PBMCs
CD4+ or CD8+ T cell subpopulations were depleted from fresh PBMC preparations, using Miltenyi Biotec (Paris, France) antibodies specific for macaque CD4+ or CD8+ T cells and MS columns according to the manufacturer's instructions. The purity of the negative subpopulations was monitored by immunolabeling with anti-CD4 and anti-CD8 antibodies (BD Biosciences). These procedures resulted in contamination of less than 9% for CD4 and 2% for CD8 as assessed by flow cytometry.
Intracellular cytokine staining
Fifteen-milliliter conical tubes were coated with 1 ml of goat anti-mouse antibody at 2.5 μg/ml in 50 mM Tris, pH 8.6, for 1 hr at 37°C. Tubes were washed twice with 1 ml of PBS and then incubated with anti-CD28 and anti-CD49b antibodies (5 μg/ml in PBS) overnight at 4°C. After two washes with PBS, 1 ml of culture medium containing 2 × 106 PBMCs was added to each tube together with 5 μg of antigen (peptides or staphylococcal enterotoxin B [SEB] plus staphylococcal enterotoxin A [SEA]). The samples were incubated for 1 hr at 37°C, in a humidified atmosphere containing 5% CO2, and 10 μg of brefeldin A (Sigma-Aldrich) was added to each tube and incubation continued for an additional 5 hr. Aliquots of 100 μl of 20 mM EDTA in PBS were added, vortexed vigorously, and the samples were incubated at room temperature for 10 min. After one wash with PBS, 1 ml of FACS lysing solution (BD Biosciences) was added to each tube, mixed, and incubated at room temperature for 10 min. Cells were washed with PBS and then incubated with 1 ml of FACS permeabilizing solution (BD Biosciences) for 10 min at room temperature. Cells were washed with PBS and then incubated with the following antibodies: IFN-γ–FITC, CD69–PE, CD4–PerCP, IL2–APC, and CD3–Alexa Fluor 700 or the corresponding isotype controls (all antibodies were from BD Biosciences), for 20 min at room temperature. Cells were washed with PBS then resuspended with 200 μl of Cell Fix solution (BD Biosciences) and analyzed with an LSR I flow cytometer (BD Biosciences).
Statistical analysis
Comparisons of data obtained from vaccinated animals versus control animals were performed by using the nonparametric Mann-Whitney test and StatView software (SAS Institute, Cary, NC).
Results
Segregation/partitioning function of the gene transport unit in dividing cells
The gene transport unit (GTU) vector we used has unique beneficial features compared with ordinary DNA plasmid vectors (Fig. 2).First, the vector expresses the nuclear anchoring E2 protein of bovine papillomavirus type 1 (BPV1) (Ilves et al., 1999), which interacts specifically through the N-terminal trans-activation domain with mitotic chromatin, tethering the plasmid through interaction of the C-terminal DNA-binding dimerization domain of the E2 protein with the multimeric E2 binding sites (Abroi et al., 2004; Djuranovic et al., 2004; Kurg et al., 2005; Zheng et al., 2005). These interactions provide segregation/partitioning function to the GTU, thereby ensuring maintenance of the plasmid in the nuclei of the dividing cell population (Abroi et al., 2004; You et al., 2004, 2005; McPhillips et al., 2005). Second, the E2 protein and E2 binding sites, in addition to the segregation/partitioning function, act strongly as a transcription enhancer (Baars et al., 2003; Kurg et al., 2006) of the promoter driving expression of the vaccine. Thus various promoters become up to 100 times stronger in the GTU context compared with regular vectors (Krohn et al., 2005). The BPV1 segregation/partitioning function was successfully transferred to the GTU by adding the BPV1 E2 expression cassette and its multimeric binding sites to the ordinary expression plasmid, as demonstrated by plasmid loss assay (Silla et al., 2005). A highly proliferating cell culture was transfected with equimolar amounts of the regular plasmid vector CMV-d1EGFP or with GTU-d1EGFP expressing the destabilized form of the enhanced green fluorescent protein (d1EGFP), or was mock transfected. Compared with CMV-d1EGFP, GTU-d1EGFP bears the same elements for expression of d1EGFP except that it also contains 10 copies of oligomerized E2 binding sites and the Rous sarcoma virus (RSV) long terminal repeat (LTR)-driven expression cassette for BPV1 E2 (Fig. 2A).
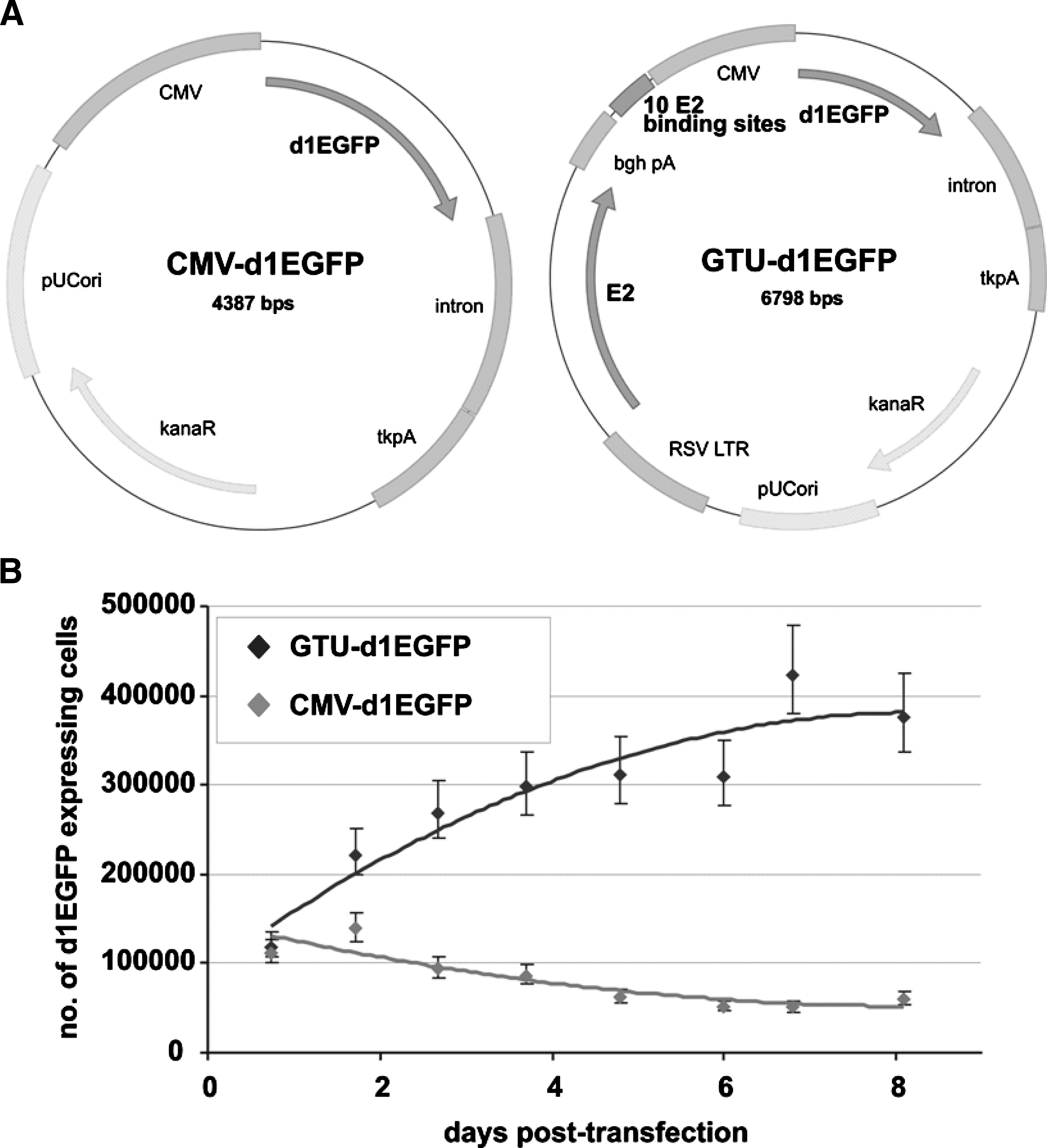
Plasmid loss assay demonstrating the segregation/partitioning function of the GTU vector. (
At various posttransfection time points, equal aliquots of cell suspension from each sample were collected for analysis and the samples were diluted thereafter with fresh medium. Changes in the total number of cells and in the number of d1EGFP-positive cells were measured by flow cytometry. As the half-life of d1EGFP is as short as 1 hr (vs. 26 hr for normal EGFP) (Li et al., 1998), the protein does not accumulate in the cells and therefore the presence of d1EGFP expression indicates the presence of the transcriptionally active vector in cells. The percentages of d1EGFP-expressing cells, alterations of total numbers of cells, and numbers of d1EGFP-expressing cells in samples were calculated; the mock-transfected sample was used as a negative control for background fluorescence.
As shown Fig. 2B, the GTU vector has segregation/partitioning capability functions fundamentally different from those of the ordinary expression vector. As predicted, the number of d1EGFP-positive cells increased over time and, on day 8, there were six times more plasmid-positive cells in the case of GTU-d1EGFP versus the regular CMV-d1EGFP vector. There was no obvious difference in total cell number in cultures transfected with either plasmid or in mock-transfected cells (data not shown). Accordingly, the increase in plasmid-positive cells was a result of plasmid segregation/partitioning into the daughter cells without any replication.
E2-dependent transcriptional activation function of the GTU
Jurkat cells and primary cell lines were transfected with equimolar amounts of ordinary CMV plasmid or GTU plasmid, using identical promoter, intron, and polyadenylation signal sequences for marker gene expression (Fig. 3). Compared with the CMV plasmid, significantly enhanced expression of either marker gene was already observed with the GTU vector 24 hr posttransfection, indicating the major effect of the E2 protein and its binding site-dependent transcriptional activation function. Expression from the GTU vector was also more long-lasting compared with the CMV plasmid, probably because of transcriptional activation and plasmid maintenance functions of the GTU vector system. These data demonstrate, by comparative expression analysis in a cell culture system with luciferase (Fig. 3A) and human vascular endothelial growth factor (VEGF)-165 (Fig. 3B) as quantitative marker genes, that the E2-dependent transcription activation function of the GTU vector system is mediated by the BPV1 E2 expression cassette and its oligomerized E2 binding sites. Similar conclusions rose from the expression of EGFP in primary cell lines obtained after ConA stimulation of macaque PBMCs and transfection with the GTU vector compared with a classical CMV plasmid (Fig. 3C).
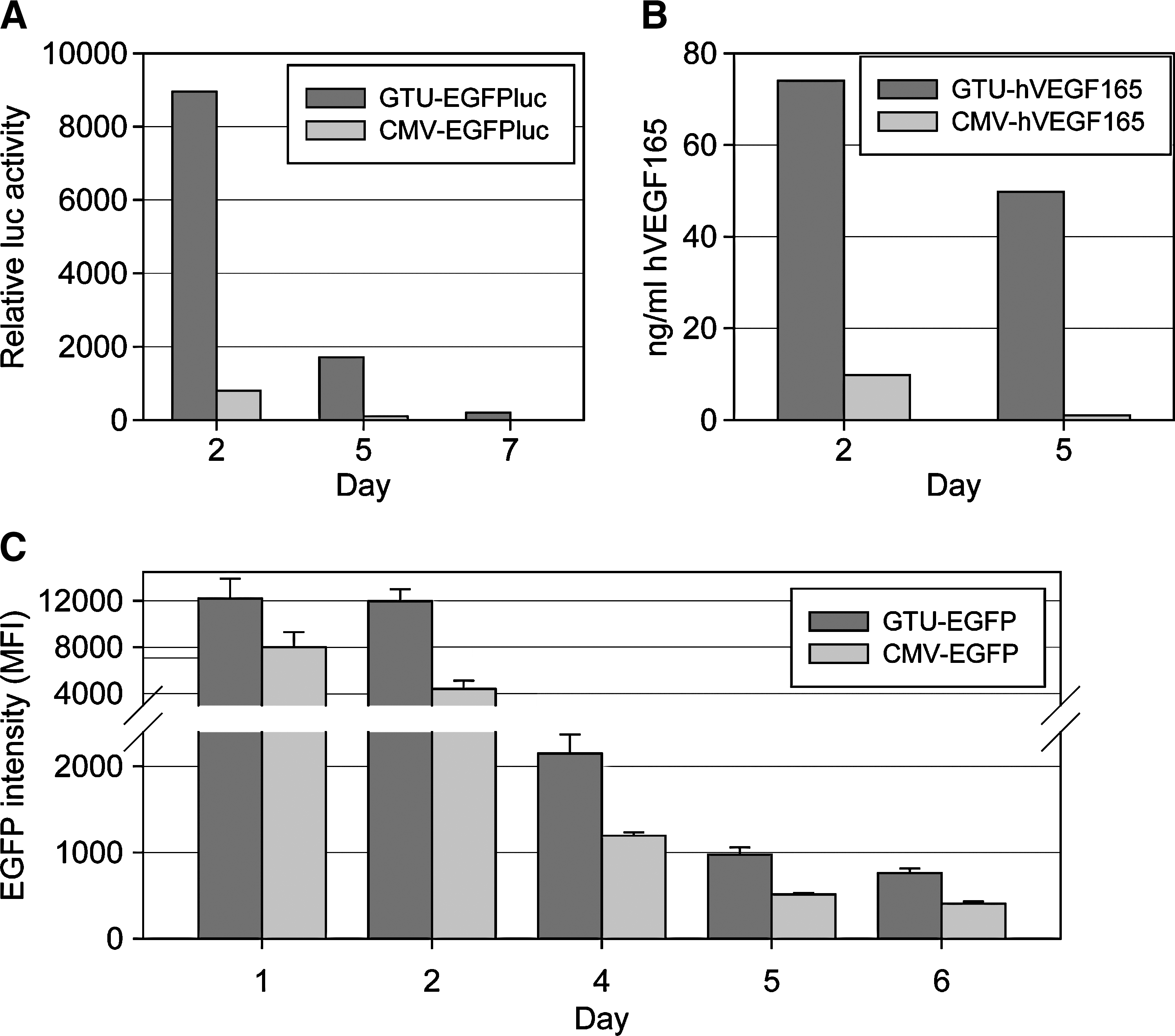
Demonstration of the E2-dependent transcription activation function exploited by the GTU vector system. Jurkat cells (
Expression of encoded antigen after intradermal delivery
We first explored intradermal delivery and antigen expression in vivo in pig skin (Fig. 4A),which is a standard biomedical animal model of human skin, especially suitable for studies of DNA uptake and gene expression (Hengge et al., 1996). Intradermal injection of swine skin with auxo-GTU-MultiHIV combined with EP resulted in significant expression of both the MultiHIV antigen and the E2 protein (Fig. 4B); both products were found mainly in the epidermis, consistent with previous findings (Hengge et al., 1996).
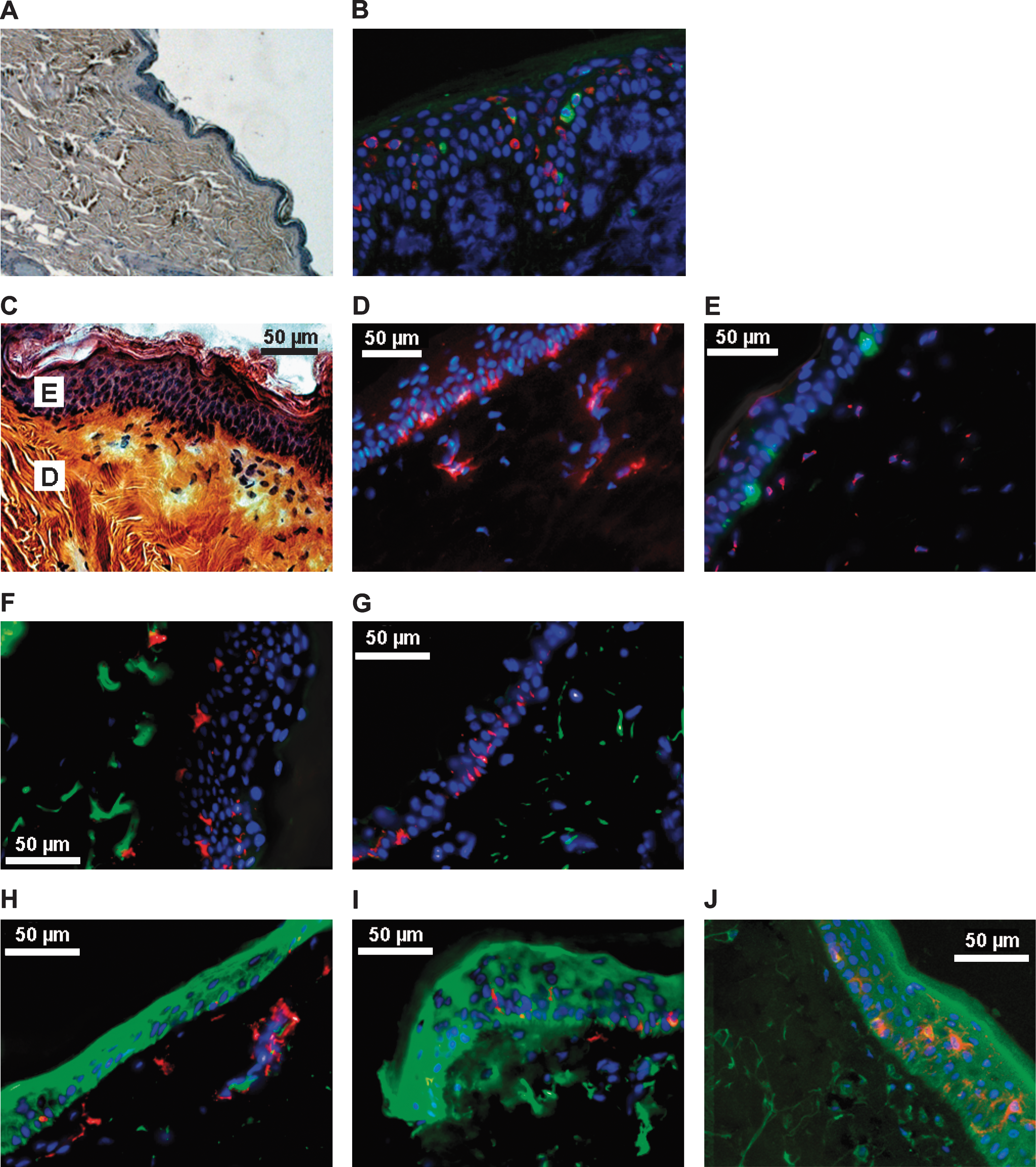
Histology of skin biopsies. Tissues were collected from pigs (

The organization of macaque skin is also similar to that of human skin (Fig. 4C), with the presence of immunocompetent cells expressing MHC class II molecules (Fig. 4D). These cells include Langerhans cells (LCs), localized in the epidermis, expressing CD1a; and interstitial dendritic cells (DCs), expressing CD209, in the dermis (Fig. 4E). Intradermal injection of macaques with the plasmid but without EP resulted in antigen expression only in the dermis (Fig. 4F and G); EP substantially increased antigen expression in the epidermis (Fig. 4H and I). p24 antigen was detected in the whole thickness of the epidermis. Stronger expression was found in the external layer of keratinocytes (Fig. 4J). Semiquantitative analyses confirm that expression of the antigen in the dermis was similar in both groups of vaccinated animals whereas the increase in the epidermis and stratum corneum, due to EP treatment, was highly statistically significant (Fig. 4K). We focused on cells expressing p24 antigen in the epidermis (Fig. 5).Confocal microscopy images showed that some TGM1+ cells, that is, more differentiated epidermal keratinocytes (Thacher and Rice, 1985), strongly expressed p24 antigen. Figure 5A shows that CD1a+ cells in contact with TGM1+ cells were also p24+ whereas, when they were close to the basal layer, the CD1a+ cells were p24−. Semiquantitative analyses demonstrated that differentiated TGM1+ keratinocytes expressed higher levels of p24 antigen than less differentiated cells of the epidermis (Fig. 5B).
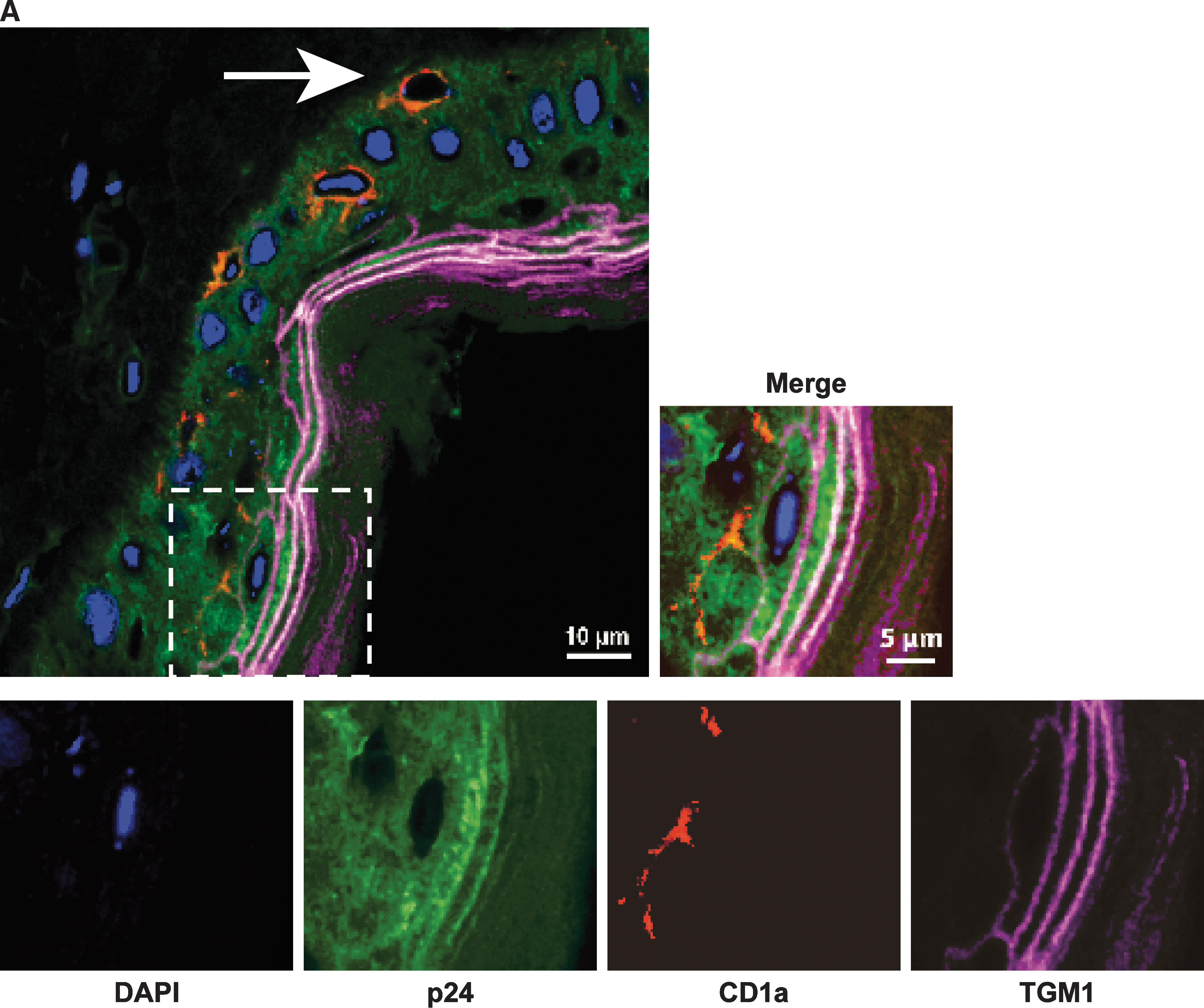
Cell types expressing antigen in macaque epidermis. (

Immune responsiveness to MultiHIV antigen in macaques
For immunological studies, two groups of four macaques received plasmid vector by two different administration methods: conventional intradermal injection (Mantoux method) and intradermal injection combined with EP. The animals received 1 mg of plasmid (100 μl at each of 10 different sites) on each of weeks 0, 4, and 12. Four additional macaques received the same vector without antigenic insert (empty vector): two controls were given intradermal injections and two were given intradermal injections associated with EP. Antibody responses were observed in the sera of both groups of vaccinated NHPs (Fig. 6A); however, the titers in non-EP animals were low and faded within 4 months of the last vaccine injection. Responses were higher in animals injected in combination with EP. The antibody responses appeared earlier in the intradermal injection plus EP group, and all four animals were seropositive before the third immunization. Only one in the non-EP group was seropositive before immunization, and another seroconverted after the third immunization. Enzyme-linked immunosorbent assay (ELISA) experiments indicated that Nef and Gag were strongly immunogenic. Although the titer of anti-Tat antibodies was more than 10-fold lower, these antibodies were able to neutralize Tat activity in vitro (Fig. 6B).
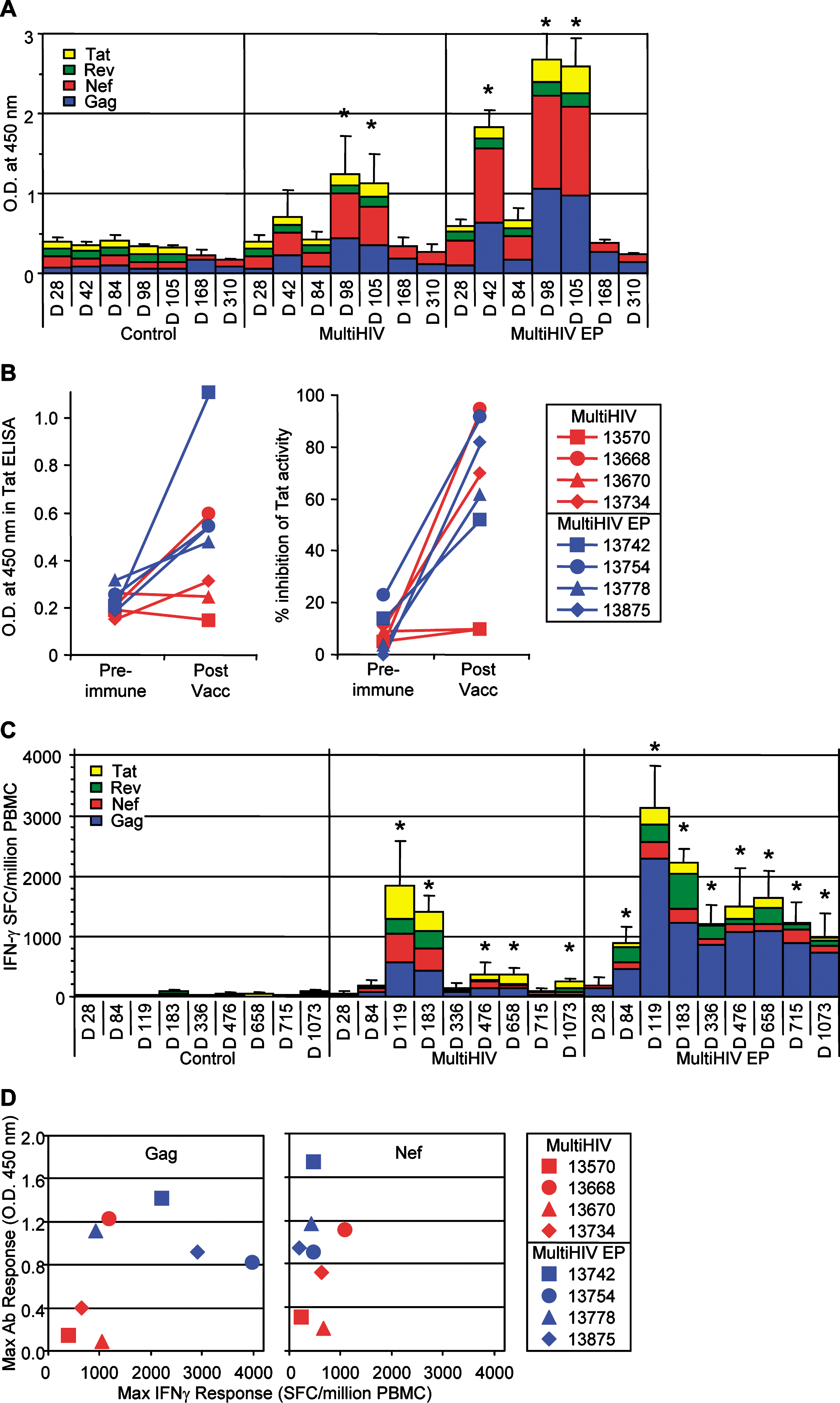
Vaccine-induced immune responses in nonhuman primates (NHPs). (
IFN-γ-producing T cells, specific for the four plasmid-encoded proteins, were detected by ELISpot assay in all the vaccinated animals, and the kinetics of the response was similar in all animals (Fig. 6C). The peak response was late, 1 month after the third injection (day 119). In the vaccinated animals without EP, the response remained high for 2 months after this peak and then decreased, although it was still significantly higher than in control animals (p = 0.0339) 33 months after the last boost (day 1073). Interestingly, intradermal injection followed by EP induced even stronger T cell responses, with a peak at 3356 ± 692 SFCs per million PBMCs (mean ± SEM of the cumulative responses for the four antigens; range, 1112–4902; n = 4). The response remained high and stable, above 1000 SFCs per million PBMCs between 11 and 33 months postvaccination. The concordances between antibody and ELISpot responses were plotted for each vaccinated animal (Fig. 6D). This analysis confirms the enhancing effect of EP on antibody responses for both Gag and Nef antigens. By contrast, only Gag-specific ELISpot responses were increased by EP. For both antigens, the correlation between responses was not statistically significant (p = 0.1577 and 0.0973 for Gag and Nef, respectively).
Individual Rev peptides were used in ELISpot assays to map epitopes recognized by animal 13778 from the EP group (Fig. 7A). Two overlapping peptides efficiently stimulated T cells, indicating that region 62–72 is the major target in the Rev protein for this animal. CD4+ or CD8+ T cell subpopulation depletion from fresh PBMCs before ELISpot assays indicated that this epitope was recognized by CD8+ T cells (Fig. 7B). A similar approach was used to identify Gag epitopes recognized by the T cells of animals 13742 and 13754 from the EP group (Fig. 7C–F). Both animals were high responders against Gag protein. Epitope mapping identified regions Gag p24 30–40 and Gag p24 175–187 as major epitopes recognized by animal 13742; and regions Gag p17 19–31 and Gag p24 30–40 for animal 13754. These Gag peptides were then used in ICS to evaluated the frequency of CD3+CD4+ cells or CD3+CD4− cells (which we assume are mainly CD8+ T cells) producing IFN-γ and IL-2 on peptide stimulation (Fig. 8A). Figure 8B shows that 1 year after the last vaccine boost (day 472 after the first immunization) activated (CD69+) cells within the CD4+ T cells were still detectable in animals of the EP group. The proportion of these cells was similar for all four, whereas CD69+ cells among the CD8+ T cells were increased for the animals demonstrating high ELISpot responses (animals 13742 and 13754) when compared with moderate responders (animals 13778 and 13875). In response to Gag peptide stimulation, CD4+ T cells mainly produced both cytokines IFN-γ and IL-2, for all four animals. By contrast, a high proportion of CD8+ T cells (CD3+CD4+) produced only IFN-γ; nevertheless, a small proportion of activated CD8+ T cells (0.5 to 6%) still produced both IL-2 and IFN-γ (Fig. 8B).
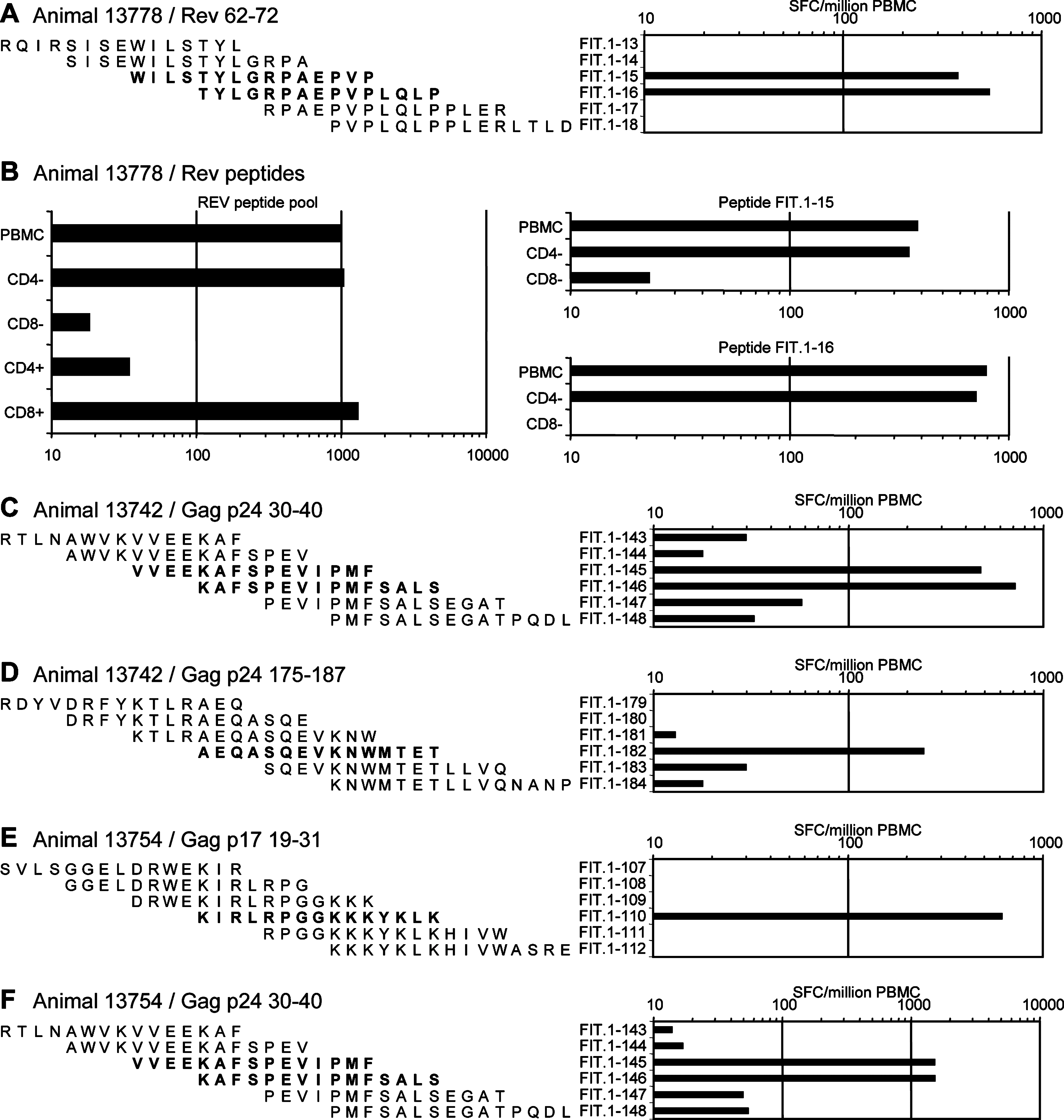
Epitope mapping of T cell responses. PBMCs from high responders of the EP group were used in ELISpot assays for the recognition of individual peptides from Rev (
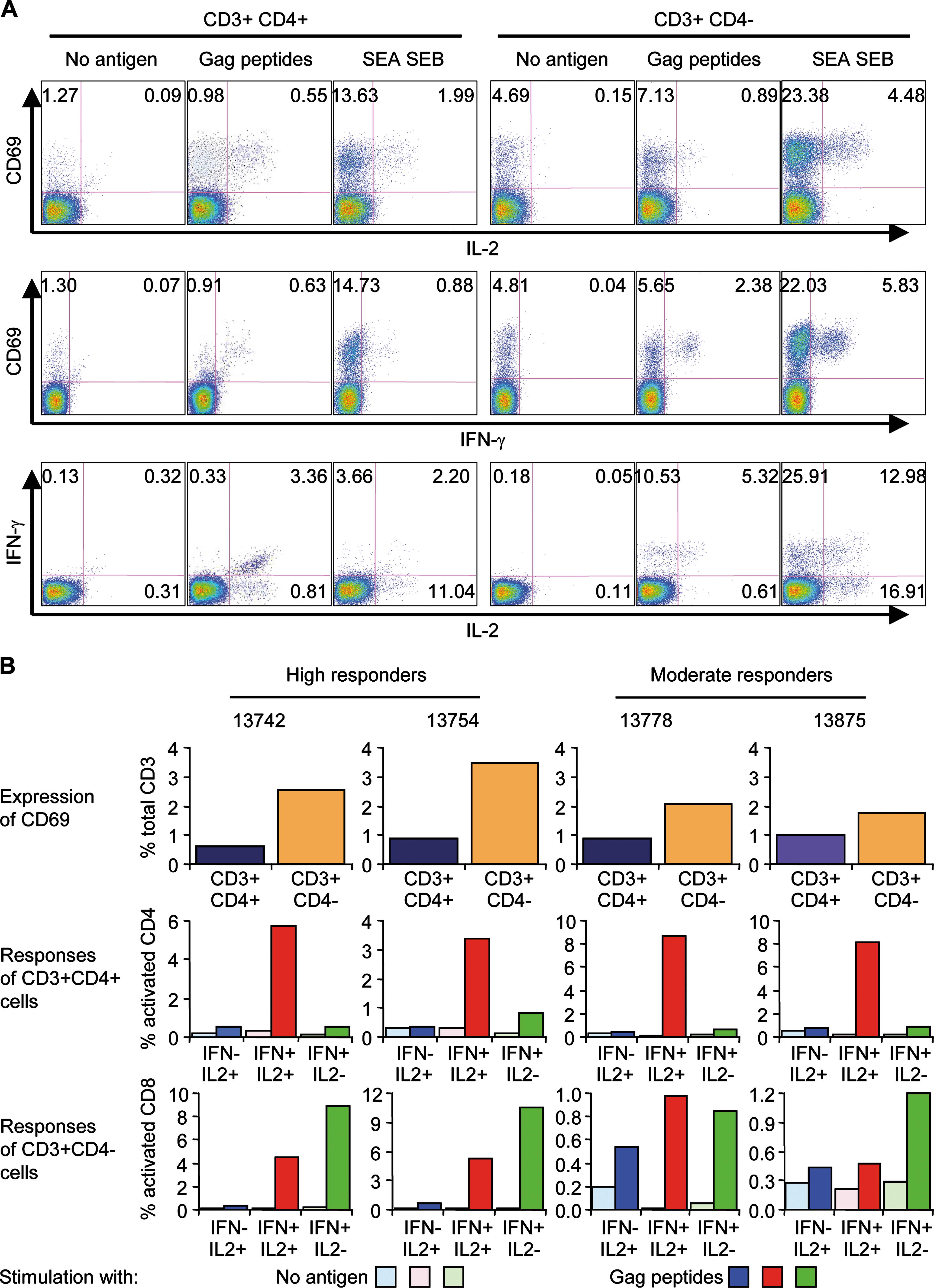
Gag-specific activation and cytokine production in vaccinated NHPs from the EP group. (
Discussion
The GTU vector is a nonreplicating plasmid expression vector with enhanced features provided by the BPV1 transcription activator and segregation/partitioning factor E2 protein along with its multimeric specific binding sites (Ilves et al., 1999). The E2 protein tethers the plasmid to chromatin through all phases of the cell cycle and serves as a strong multimeric E2-binding site-dependent transcription activator of the antigen-encoding genes. The BPV1 E2 protein, functioning through E2 binding sites in the upstream regulatory region of the viral genome, modulates transcription of the early viral genes (McBride et al., 1991). In addition, the E2 protein can activate heterologous promoters if its binding sites are cloned either upstream or downstream of the transcription initiation site (Hawley-Nelson et al., 1988; Thierry et al., 1990; Gauthier et al., 1991). It is assumed that the E2 protein stimulates transcription by direct or indirect interactions with components of the transcriptional machinery, including Sp1 (Li et al., 1991), transcription factor IID (TFIID; Rank and Lambert, 1995), transcription factor IIB (TFIIB; Rank and Lambert, 1995), and activation domain-modulating factor (AMF)-1 (Breiding et al., 1997). Also, the binding of E2 protein to its tandem binding sites can modify the chromatin structure around the promoter (Li and Botchan, 1994; Lefebvre et al., 1997). Thus, with the prospect of using the GTU vector as a vaccine or therapeutic gene vehicle, the increased number of cells expressing the gene of interest and the increased expression by the E2-dependent transcriptional activation potentially allow the effective administration dose to be reduced and the time of functioning of the therapeutic gene in the tissue to be extended.
A novel auxotrophic bacterial selection marker was used to produce the vaccine. This vector was used as a DNA vaccine delivered to the skin, especially the epidermis, which is a particularly attractive tissue for vaccine delivery. In addition to practical issues, including easy administration facilitating mass vaccination, the epidermis is a preferred target tissue due to the presence of immunocompetent cells of both the innate and adaptive immune systems. Epithelial cells produce a wide array of antimicrobial peptides as well as proteins and constitute the first line of innate defense against invading microorganisms (Schroder and Harder, 2006).
Our aim was to develop a vaccine for intradermal injection that elicits a strong CD8+ T cell-mediated immune response. EP has been previously evaluated in vivo in association with intradermal and intramuscular delivery of therapeutic agents (Mir et al., 1998; Sersa et al., 2000) and with plasmid DNA (Prud'homme et al., 2006; Luckay et al., 2007; Hirao et al., 2008b). We used topical EP with needle-free tweezer-type electrodes pinching the skin at the site of vaccine injection to maximize the number of cells expressing the vaccine antigen.
NHPs, particularly macaques, are good models of the human immune system, allowing prediction of immunogenicity and, when relevant challenge is possible, identification of correlates of protection (Staprans and Feinberg, 2004). In the untreated skin of NHPs, the LCs (CD1a+) are located close to the epidermal basal layer (Fig. 4E). A similar location was observed for LCs in the skin of non-EP NHPs (Fig. 4G) whereas, after EP, some of the LCs were clearly shifted toward the external layer of keratinocytes, which strongly expressed the p24 antigen (Fig. 4J). In addition, confocal microscopy analysis indicated that LCs close to the basal layer were p24− whereas they became p24+ in the vicinity of differentiated keratinocytes that strongly expressed the antigen (Fig. 5). Although we cannot exclude the direct transfection of particular LCs, these observations suggest the attraction of LCs by the source of antigen production in the epidermis. Although further analyses would be required to understand why the differentiated keratinocytes strongly produce the antigen, the localization of LCs in the epidermis may be particularly favorable for antigen uptake by these cells, leading to the stimulation of CD8+ specific T cells (Klechevsky et al., 2008).
Our immunization strategy led to an exceptionally high and long-lasting specific T cell response in macaques, and this has never previously been described to occur against HIV antigens. In both groups of vaccinated animals, and particularly in the EP group, the T cell responses were remarkably long-lasting relative to previous reports concerning DNA vaccines: This is the first example of an immune response, without repeated intermediate boosts, sufficiently persistent for prophylactic vaccination. The response was also greater than those reported for prime–boost strategies combining DNA-based vaccines and recombinant viral vector (Casimiro et al., 2005).
Presumably, the epidermal delivery and the characteristics of the auxo-GTU DNA vaccine combined to produce the immunization effects observed. EP substantially increased the magnitude of both early and late immune responses. The exceptionally long persistence of the specific responses may result from the generation of large numbers of specific T cells during the expansion period, the prolonged production of the antigens, or both. In addition, local inflammation of the skin due to EP may favor stimulation of the immune system and may enhance the generation of an adaptive response.
In the non-EP group, the specificity of the T cell responses was equally distributed against Gag, Nef, and Tat proteins, and the responses to Rev were lower in ELISpot assays. In the EP group, the largest increase in T cell responses was directed to Gag (Fig. 6). Because the HIV genes included in the plasmid were produced as a fusion protein (Blazevic et al., 2006), their relative expression should be equivalent. Therefore, the dominance of anti-Gag T cell responses would therefore be due to the immune properties of this antigen (Goulder et al., 1997; Matano et al., 2004) rather than to the level of production in vivo. The fact that the level of antibodies to Gag did not correlate with the level of anti-Gag T cell responses (Fig. 6) argues in favor of this hypothesis. Lack of correlation was also observed for anti-Nef responses; nevertheless, in this particular case it seemed to be due to the small number of animals in the experiments. In addition, because the MultiHIV antigen was produced as a fusion protein, anti-Gag T cell responses could provide the help required for anti-Nef antibody responses (Rajewsky et al., 1969). One animal also had large numbers of IFN-γ-producing cells specific to Rev. Thus, injection combined with EP favored the development of anti-Gag T cell responses, at least with the particular vaccine used. Interestingly, strong and broad anti-Gag responses are associated with low viremia in HIV-infected patients (Kiepiela et al., 2007). Epitope mapping indicated that the strongest IFN-γ responses were mediated by CD8+ T cells and were directed toward Rev and Gag peptides (Fig. 7) included in epitope-enriched regions for humans (see
To conclude, we have succeeded in inducing a strong and durable T cell response in macaques by using a DNA-based strategy for immunization with a DNA vaccine encoding an artificial multicomponent fusion antigen consisting of several proteins or protein fragments of HIV-1. This response was still detectable 33 months after the last vaccination. The strength of the CD8+ T cell response seems to be associated with expression of the antigen in the epidermal compartment. The induction of such a robust T cell response with a DNA vaccine alone is of particular interest in the development of vaccine strategies. We also demonstrated that an antigen designed to induce potent cellular immunity to multiple viral antigens can be effective. MultiHIV is an artificial fusion protein consisting of several HIV-1 proteins and protein fragments. Alternative approaches in multivalent vaccine development exploit separate vectors, each encoding different antigens. However, single-vector vaccines are clearly more practical and both easier and less expensive to produce than multiple-vector vaccines. We show here that immunization of macaques with a single vector encoding the large multicomponent antigen induced a long-lasting cellular immune response against all constituents of the antigen. Whether the induction of T cell responses is sufficient to provide protection against HIV is currently being explored in phase IIa clinical trials.
Footnotes
Acknowledgments
The authors warmly thank D. Renault, P. Pochard, J.C. Wilk, C. Jouy, P. Flammant, H. Juin, and J.C. Mascaro for animal care and the veterinary staff under the supervision of C. Joubert. The authors thank Dr. B. Verrier for providing the anti-p24 monoclonal antibody. The authors thank Dr. D. Vignjević for assistance in image analyses. This work was supported by the AIDS Vaccine Integrated Project (AVIP; European Commission, grant LSHP-CT-2004-503487), EPIVAC (European Commission, grant LSHP-CT-2006-037651), and Europrise (European Commission, grant LSHP-CT-2006-037611).
Author Disclosure Statement
No competing financial interests exist.