
Abstract
In vivo gene transfer to the human respiratory tract by adenovirus serotype 5 (Ad5) vectors has revealed their limitations related to inefficient gene transfer, host antiviral response, and innate adenoviral toxicity. In the present work, we compared the cytotoxicity and efficiency of Ad5 and a chimeric Ad5F35 vector with respect to CFTR gene transfer to cystic fibrosis (CF) and non-CF human airway epithelial cells. We found that high doses of Ad5 vector had an adverse effect on the function of exogenous and endogenous CFTR. Results obtained with Ad5 capsid mutants suggested that the RGD motifs on the penton base capsomers were responsible for the negative effect on CFTR function. This negative interference did not result from a lower level of biosynthesis and/or altered cellular trafficking of the CFTR protein, but rather from an indirect mechanism of functional blockage of CFTR, related to the RGD integrin-mediated endocytic pathway of Ad5. No negative interference with CFTR was observed for Ad5F35, an Ad5-based vector pseudotyped with fibers from Ad35, a serotype that uses another cell entry pathway. In vitro, Ad5F35 vector expressing the GFP-tagged CFTR (Ad5F35-GFP-CFTR) showed a 30-fold higher efficiency of transduction and chloride channel correction in CFTR-deficient cells, compared with Ad5GFP-CFTR. Ex vivo, Ad5F35-GFP-CFTR had the capacity to transduce efficiently reconstituted airway epithelia from patients with CF (CF-HAE) via the apical surface, restored chloride channel function at relatively low vector doses, and showed relatively stable expression of GFP-CFTR for several weeks.
Introduction
Adenoviruses in general, and Ad5 in particular, had been used as gene transfer vectors for more than a decade, but the limitations of this vector were revealed during attempts to deliver cDNA encoding the cystic fibrosis transmembrane conductance regulator (CFTR) to differentiated airway epithelia for the correction of the cystic fibrosis defect (Bradbury, 1999). Besides host antiviral response and innate adenoviral toxicity, one of the major reasons for the inefficient Ad5-mediated gene transfer to the airways was the topology of CAR in the airway epithelium. CAR is inaccessible to Ad5 vectors, as it is localized beneath the tight junctions on the basolateral membrane (Walters et al., 1999, 2002).
It has been reported that pseudotyping Ad5 with serotype 35 fibers redirects the chimeric Ad5F35 vector to CD46 molecules, and to heparan sulfate proteoglycans, which also act as adenoviral receptors (Gaggar et al., 2003; Wu et al., 2004; Tuve et al., 2008). Normal human airways have been shown to express CD46 molecules at their apical surface (Sinn et al., 2002), leading to the hypothesis that Ad35 or Ad5F35 chimera would be more efficient vectors for airway epithelia. Ad5F35 was found to transduce polarized human intestinal epithelium via the apical pole, and, among a panel of various fiber-pseudotyped Ad5 vectors, it was the most efficient transducer through this pathway (Lecollinet et al., 2006). Ad5F35 has also been successfully used as an oncolytic vector in many types of malignancies (Hoffmann et al., 2007; Toyoda et al., 2008; Wang et al., 2009). In addition, Ad5F35 chimeric vectors show a better safety profile after intravenous injection into CD46-transgenic mice than do Ad5 vectors (Ganesh et al., 2009). More recently, it was found that the chimeric Ad5F35 vector preferentially localized to the lungs of CD46-transgenic mice at higher levels compared with Ad5 (Greig et al., 2009), suggesting that chimeric Ad5F35 vectors have a great potential for gene transfer to lung tissue in vivo. Interestingly, pseudotyping an Ad2 vector with serotype 17 fibers enhanced the gene transfer efficiency in human airway epithelia (Walters et al., 1999; Zabner et al., 1999). Of note, Ad17 belongs to subgroup D adenoviruses, characterized by short fibers (13 nm in length), a length similar to that of fibers of Ad35 (11 nm).
In this paper, the vectors Ad5 and Ad5F35, both carrying the green fluorescent protein (GFP)-tagged CFTR gene, were compared in terms of transduction efficiency and CFTR chloride channel activity after gene delivery to cystic fibrosis (CF) and non-CF human airway epithelial cells. We found that transduction with the Ad5 vector at high vector doses had a negative effect on the activity of endogenous CFTR in normal human tracheal glandular serous cells. Such an inhibition of a plasma membrane-resident protein function by a viral vector has not been previously described. A similar negative effect was observed on exogenous CFTR expressed in CFTR-deficient cells transduced with high vector doses of Ad5GFP-CFTR. The present study provides experimental evidence that the negative interference of Ad5 with the CFTR chloride channel function was associated with the RGD motifs of the penton base and the RGD–integrin-mediated endocytic pathway. Interestingly, Ad5F35 had no adverse effect on the CFTR function. Transduction of well-differentiated human airway epithelium from CF patients with an Ad5F35-GFP-CFTR vector showed that Ad5F35 entered epithelial cells via their apical pole and corrected the chloride channel defect at significantly lower vector doses, compared with those required with the Ad5GFP-CFTR vector. Pseudotyping Ad5 with serotype 35 fibers therefore conferred a significant advantage to the chimeric adenoviral vector over conventional serotype 5 vectors, in terms of transduction of airway epithelia in vivo and ex vivo.
Materials and Methods
Cell lines
Human tracheal gland serous cells (MM-39) were from the tracheal mucosa of a young healthy adult and CF-KM4 are serous cells from a patient with CF carrying a ΔF508-ΔF508 mutation (Merten et al., 1996; Kammouni et al., 1999; Gaden et al., 2002, 2004). Both cell lines were simian virus 40 (SV40) transformed. MM-39 and CF-KM4 cells were maintained as monolayers in Dulbecco's modified Eagle's medium (DMEM)–Ham's F12 supplemented with 1% Ultroser G (GIBCO/Invitrogen, Carlsbad, CA), sodium pyruvate (1 mM), glutamine (5 mM), penicillin (200 U/ml), streptomycin (200 μg/ml), and epinephrine (3 μM), on collagen I-coated flasks (BioCoat; BD Biosciences, San Jose, CA). A549 and HEK-293 cells, obtained from the American Type Culture Collection (ATCC, Manassas, VA), were maintained as monolayers in DMEM (GIBCO/Invitrogen) supplemented with 10% fetal bovine serum (FBS; GIBCO/Invitrogen), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C, 5% CO2.
Primary human airway epithelial cell culture model
Cells from human airway epithelia (HAE) were isolated from tracheas and bronchi of donor lungs. Cells were seeded onto collagen-coated, semipermeable membranes (0.6 cm2 Millicell-HA; Millipore, Bedford, MA), and grown at an air–liquid interface as previously described (Zabner et al., 1996, 2005). At 36–48 hr after seeding, the cells form a confluent culture with electrically tight junctions, and between days 3 and 14, the epithelial cells differentiate into a predominantly ciliated phenotype (Zabner et al., 2005). HAE and CFTR-deficient CF HAE cells were cultured in a 1:1 mixture of DMEM and Ham's medium supplemented with 2% Ultroser G, penicillin (100 U/ml), streptomycin (100 μg/ml), gentamicin (10 μg/ml), colimycin (25 μg/ml), and ceftazidime (75 μg/ml), imipenem (25 μg/ml), cilastatin (25 μg/ml), and fluconazole (2 μg/ml). Basolateral culture medium was changed every 2–4 days. Samples were collected with approval from the University of Iowa (Iowa City, IA) Institutional Review Board.
Adenoviral transfer vectors
The Ad5GFP-CFTR recombinant vector, encoding the wild-type allele of the CFTR gene fused to the 3′ end of the GFP gene, has been described in a previous study (Granio et al., 2007). Capsid-modified adenoviral vectors were generated by homologous recombination, as described in detail in previous studies (Magnusson et al., 2001, 2002; Henning et al., 2002; Hong et al., 2003). Chimeric Ad5F35-GFP is a GFP-expressing vector carrying the shaft and knob domains of serotype 35 fiber (F35) fused to the Ad5 fiber tail, and reinserted into the E1-deleted Ad5GFP genome in place of the Ad5 fiber gene. Ad5F35-GFP-CFTR was constructed by reinsertion of the chimeric 5F35 fiber gene into the genome backbone of Ad5GFP-CFTR. Ad5F35-GFP-CFTR expresses a GFP-tagged version of the CFTR protein. Ad5PbEGDGFP, encoding GFP and carrying an RGD-to-EGD substitution at position 340 in the penton base coding sequence, has been described elsewhere (Waszak et al., 2007). The two fiber knob-deleted recombinants Ad5GFPΔknob and Ad5GFP-R7-RGD4C have been characterized in previous studies (Magnusson et al., 2001; Henning et al., 2006; Franqueville et al., 2008). Both are GFP-expressing vectors carrying a short-shafted fiber with seven repeats (R7) and complete deletion of the fiber knob domain, as in Ad5GFPΔknob, or replacement of the knob by a disulfide bond-constrained RGD ligand (ACDC
Soluble adenoviral proteins and empty particles
Adenoviral structural proteins used in the present study were isolated from two different sources: (1) Penton base (Pb), wild-type and RGD mutant (PbEGD), and fiber knob domain were expressed as recombinant proteins, and isolated from Autographa californica multicapsid nuclear polyhedrosis virus (AcMNPV)-infected Sf9 cell lysates (Novelli and Boulanger, 1991; Karayan et al., 1994, 1997; Hong et al., 2005); and (2) the other capsid proteins, hexon, fiber, and vertex capsomer penton (penton base and fiber), were recovered from the pool of soluble antigens in Ad5-infected HEK-293 cell lysates (Boulanger and Puvion, 1973). Adenoviral proteins were purified in a three-step procedure previously described (Boulanger and Puvion, 1973; Molinier-Frenkel et al., 2002), with the following modifications: (1) Adenoviral proteins were first recovered from clarified infected cell lysates by ammonium sulfate precipitation (55% saturation, pH 6.5); (2) the protein pellet was resuspended in 50 mM sodium phosphate buffer, pH 6.8 (NPB-50), and subjected to anion-exchange chromatography in a high-performance liquid chromatography system (BioLogic DuoFlow; Bio-Rad) and a DEAE-Sepharose Fast Flow column (DFF-100; Sigma-Aldrich, St. Louis, MO) equilibrated in NPB-50. Protein samples (2 to 3 mg) were applied to the column, and elution was performed by applying a 0.0 to 0.6 M NaCl gradient in NPB-50. Fiber protein was eluted within 180–200 mM salt, penton and penton base were eluted at 250 mM salt, and hexon was eluted at 425 mM salt; and (3) proteins were then further purified by adsorption chromatography on a hydroxyapatite column (Econo-Pac CHT-II; Bio-Rad) equilibrated in 10 mM sodium phosphate buffer, pH 6.9 (NPB-10). Protein samples, dialyzed against NPB-10, were loaded on the column, and a sodium phosphate gradient (10 to 300 mM) was applied to the column. Penton and penton base proteins were eluted at 90–100 mM sodium phosphate, hexon at 125–150 mM, and fiber at 200–220 mM. When required, proteins were concentrated with concentrator membranes with a 100-kDa cutoff (Vivaspin-100; Vivascience, Binbrook, UK). Protein concentration was estimated by Bradford assay (Bio-Rad). Proteins were analyzed by conventional sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting, as previously described (Karayan et al., 1994, 1997; Hong et al., 2005). Empty adenoviral particles devoid of genomic DNA, referred to as top components (D'Halluin, 1995), were recovered from CsCl gradient ultracentrifugation used for the preparation of wild-type adenoviral stocks (Defer et al., 1990). Their concentration was determined by Bradford assay, and their titer expressed as physical particles per milliliter, as previously mentioned. Detergent disruption of adenoviral particles was performed by heating virion samples at 56°C for 90 sec in low-salt buffer (5 mM) containing 0.5% sodium deoxycholate (DOC) (Boulanger et al., 1977), and then the DOC was eliminated by dialysis.
Fluorescence microscopy
Cell monolayers or epithelia were infected with recombinant adenovirus at various multiplicities of infection (MOIs), ranging from 25 to 5000 VP/cell, corresponding to infectious titers of about 1 to 200 PFU/cell for Ad5 vectors, as titrated in HEK-293 cells. After 1 hr of viral adsorption at 37°C, the cells were rinsed with prewarmed serum-free medium and further incubated for 48 hr with complete medium at 37°C. Cells were observed directly for GFP expression, after incubation with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining. Direct observations were made with a Zeiss Axiovert 135 inverted microscope equipped with an AxioCam digital camera (Carl Zeiss, Oberkochen, Germany). For confocal fluorescence microscopy, epithelia were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 and 2% BSA in SuperBlock (Pierce Biotechnology, Rockford, IL). After blocking with 2% BSA in SuperBlock for 1 hr at room temperature, cells were incubated with primary antibody (mouse anti-human ZO-1 antibody, diluted 1:250; BD Biosciences) for 3 hr at 37°C, rinsed in phosphate-buffered saline (PBS), and then incubated with fluorophore-conjugated secondary antibody (Alexa Fluor 568-conjugated goat anti-mouse IgG antibody, diluted 1:1000; Invitrogen) for 90 min at 37°C. After several rinses, cells were mounted on glass slides and a coverslip was placed with VECTASHIELD plus DAPI mounting solution (Vector Laboratories, Burlingame, CA). Observations were performed with a Leica TCS SP2 confocal microscope (Leica Microsystems, Wetzlar, Germany) or an Olympus FluoView FV1000 laser scanning confocal microscope (Olympus, Tokyo, Japan) equipped with a ×60 oil immersion lens, and analysis was performed with Olympus FluoView viewer software version 1.4a and ImageJ (National Institutes of Health, Bethesda, MD). For live-cell imaging, images were acquired with an IX81 motorized Olympus inverted microscope, piloted by Volocity 3D-image processing software (Improvision/PerkinElmer Life and Analytical Sciences, Waltham, MA).
Quantification of adenoviral vector genomes and CFTR mRNA molecules by real-time PCR and real-time RT-PCR
Quantification of the cell-internalized adenoviral genome copy number in vector-transduced cells was performed by real-time polymerase chain reaction (PCR) using human adenovirus-specific primers and probe selected from the 3′ end of the hexon gene (Van Tol et al., 2005) with minor modifications. The extraction procedure was carried out with a High Pure viral nucleic acid kit (Roche Diagnostics, Mann-heim, Germany) and quantitative PCRs were carried out with a TaqMan PCR reagent kit (Applied Biosystems, Foster City, CA) and the ABI PRISM 7700 sequence detector (Applied Biosystems), as previously described (Najioullah et al., 2001). For quantification of the amount of exogenous CFTR mRNA transcribed from the adenoviral vector and differentiation from endogenous CFTR mRNA, total RNA was extracted with a QiaAmp viral RNA mini kit (Qiagen, Hilden, Germany), and the reverse transcription (RT) reaction was performed with an antisense primer designed from the 5′ end of the CFTR gene (nucleotide position 78 in the CFTR gene; 5′-GCGCTGTCTGTATCCTTTCCTCAA) and avian myeloblastosis virus (AMV) reverse transcriptase (Promega, Madison, WI) for 1 hr at 37°C followed by 5 min at 95°C to inactivate the enzyme. Quantitative real-time PCR was performed with SYBR green master mix 2 × (Applied Biosystems) and the 7500 real-time PCR system (Applied Biosystems), with a forward primer designed from the 3′ end of the GFP gene (nucleotide position 621; 5′-AACGAGAAGCGCGATCACATG) and the reverse primer at position 78 in the CFTR gene. The PCR-amplified fragment was 196 nucleotides in length and overlapped the GFP and CFTR junction sequence.
Apical transduction of airway epithelia
Apical and basolateral infections were performed as previously described (Walters et al., 1999; Zabner et al., 1999; Granio et al., 2007). Briefly, for apical transduction, 50 μl containing the recombinant virus in PBS was added to the apical surface for 1 hr, after which the viral suspension was removed and the monolayers were rinsed twice with PBS. For basolateral infection, epithelia were inverted and 25 μl of virus in PBS was adsorbed to the basolateral surface at room temperature for 30 min, rinsed, and placed in fresh medium. After infection, the epithelia were incubated at 37°C for an additional 3 to 4 days.
Analysis of CFTR channel activity by radiotracer efflux method
Cells were cultured in 24-multiwell plates and analyzed for CFTR channel activity 48 and 96 hr after infection with adenoviral vectors. The CFTR Cl− channel activity was assayed by measuring the rate of iodide (125I) efflux from cells as previously described (Derand et al., 2001; Dormer et al., 2001). All experiments were performed with a MultiPROBE II EX robotic liquid handling system (PerkinElmer, Courtaboeuf, France). At the start of each experiment, cells were washed twice with efflux buffer (136.9 mM NaCl, 5.4 mM KCl, 0.3 mM KH2PO4, 0.3 mM NaH2PO4, 4.2 mM NaHCO3, 0.5 mM MgCl2, 1.3 mM CaCl2, 0.4 mM MgSO4, 5.6 mM glucose, and 10 mM HEPES, pH 7.4). The cells were then incubated in efflux buffer containing Na125I (1 mCi of Na125I/ml; NEN/PerkinElmer Life and Analytical Sciences) for 1 hr, to allow the 125I to reach equilibrium. At the end of the incubation period, the medium was removed and cells were briefly washed with efflux buffer to remove extracellular 125I. The progressive loss of intracellular 125I was then determined by removing the medium and replacing it with fresh efflux buffer every minute for 10 min. The first three aliquots were used to establish a stable baseline of efflux buffer alone. Medium containing the appropriate drug was used for the remaining aliquots. The residual radioactivity was extracted with 0.1% SDS–0.1 N NaOH, and determined with a Packard COBRA II gamma counter (PerkinElmer). The fraction of initial intracellular 125I lost at each time point was measured and the time-dependent rates of 125I efflux were calculated according to the formula ln(125I t 1 /125I t 2 )/(t 1 − t 2), where 125I t is the intracellular 125I at time t, and t 1 and t 2 are successive time points (Venglarik et al., 1990). Curves were constructed by plotting the rate of 125I versus time. All comparisons were based on maximal values for the time-dependent rates (k = peak rates, min–1) excluding the points used to establish the baseline (k peak − k basal, min–1) (Venglarik et al., 1990; Derand et al., 2001; Dormer et al., 2001).
Assay for transepithelial ion transport
The short-circuit current (I sc) was measured in modified Ussing chambers (Jim's Instruments, Iowa City, IA) as previously described (Ostedgaard et al., 2003). Primary human airway epithelia were treated with forskolin (10–5 M) and 3-isobutyl-2-methylxanthine (IBMX, 10–4 M) for 18–24 hr before transfer to Ussing chambers to minimize basal CFTR current. For conditions with symmetrical Cl− concentrations, solutions on both surfaces of the epithelia contained (in mM): 135 NaCl, 5 HEPES, 1.2 MgCl2, 1.2 CaCl2, 2.4 K2HPO4, 0.6 KH2PO4, and 5 dextrose and were gassed with 100% O2. To create a Cl− concentration gradient, NaCl was replaced with sodium gluconate on the apical side to give a Cl− concentration of 4.8 mM. Transepithelial voltage was clamped to zero. After measuring baseline current, the following reagents were added sequentially: (1) apical amiloride (10–4 M), which inhibits apical Na+ channels and hyperpolarizes the apical membrane, thereby generating a driving force for a Cl− secretory I sc; (2) apical 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS, 10–4 M), which inhibits non-CFTR Cl− channels; (3) apical forskolin (10–5 M) and IBMX (10–4 M), which increases the cellular levels of cAMP leading to phosphorylation of CFTR and subsequent activation; and (4) apical bumetanide, which inhibits all Cl− transport. All experiments were performed in triplicate.
Statistics
Results are expressed as means ± SEM of n observations. Sets of data were compared by analysis of variance (ANOVA) or Student t test. Differences were considered statistically significant when p < 0.05. Symbols used in the figures are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, with NS indicating no significant difference. All statistical tests were performed with GraphPad Prism version 4.0 for Windows (GraphPad Software, San Diego, CA).
Results
Negative interference of Ad5GFP-CFTR at high vector input with the CFTR chloride (Cl−) channel function
It was previously shown that Ad5GFP-CFTR at infectious doses of 40–60 PFU/cell, corresponding to 1000–1500 physical particles or VP/cell, was capable of correcting CFTR Cl− channel activity in the CFTR-deficient human tracheal glandular cell line CF-KM4 (Granio et al., 2007). However, at higher MOIs (≥2500 VP/cell), an inhibition of Cl− channel activity was observed (Granio et al., 2007), implying that administration of Ad5GFP-CFTR at high doses was detrimental to the function of exogenous CFTR delivered by the vector. We therefore asked whether this negative effect involved both exogenous and endogenous CFTR, and whether it occurred at the cellular or vector level.
Use of the Ad5GFP-CFTR vector had the advantage that transduced cells could be directly analyzed by flow cytometry and that CFTR cellular trafficking could be tracked in situ by its GFP tag (Granio et al., 2007). In CF-KM4 cells infected with Ad5GFP-CFTR and analyzed 48 hr postinfection, we observed a linear dose response of the fluorescent GFP-CFTR signal within the MOI range of 500–1500 VP/cell, followed by a plateau at higher vector doses (Fig. 1a). At 96 hr postinfection, the fluorescence intensity decreased at high MOI, with a 15–20% decrease at an MOI of 2500 (Fig. 1a). High Ad5 vector doses therefore provoked a slight negative effect on CFTR protein synthesis or/and stability, but this inadequately accounted for the strong inhibition of the Cl− channel function previously observed (Granio et al., 2007).
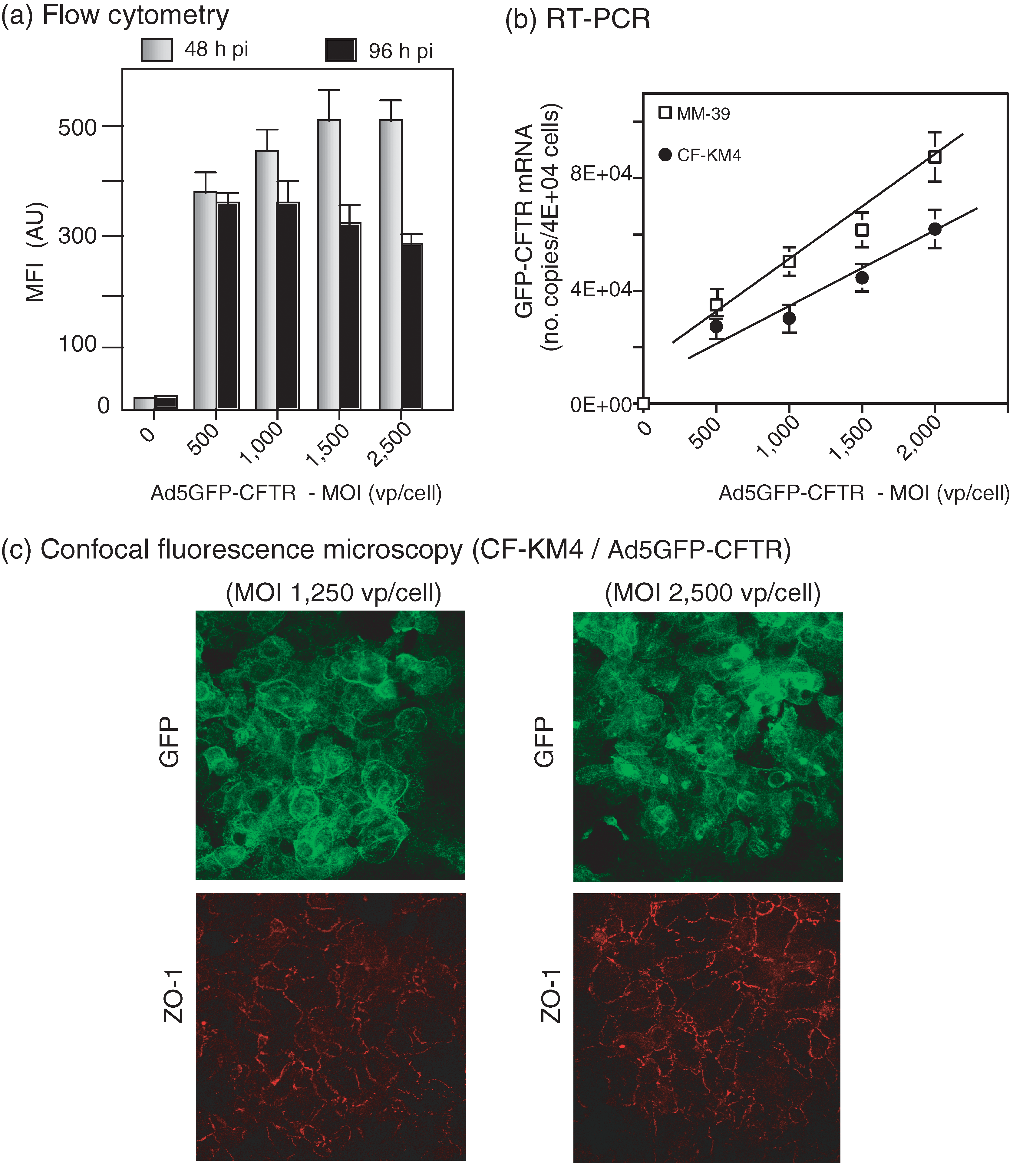
Expression of the GFP-CFTR fusion protein in CF-KM4 cells transduced by Ad5GFP-CFTR at high vector doses. (
To determine whether a transcriptional or posttranscriptional step was involved in the Ad5-induced inhibitory activity, the level of newly transcribed CFTR mRNA in Ad5GFP-CFTR-transduced CF-KM4 cells was assayed by quantitative RT-PCR 48 hr postinfection, using primers overlapping the GFP and CFTR sequences. We found a progressive, vector dose-dependent increase in the copy number of GFP-CFTR transcripts within the MOI range of 500 to 2500 VP/cell (Fig. 1b), suggesting that GFP-CFTR transcription was not affected by vector doses up to 2500 VP/cell.
We then explored the possibility of an aberrant localization of CFTR in cells transduced with a high MOI of Ad5GFP-CFTR, as previously observed in cells overexpressing CFTR (Farmen et al., 2005). CF-KM4 cells were incubated with Ad5GFP-CFTR at low and high vector doses, and analyzed by confocal fluorescence microscopy 48 hr postinfection. At the vector dose that fully restored CFTR function (MOI, 1250 VP/cell), the GFP-CFTR signal was localized at the plasma membrane, and also delineated a thin network within the cytoplasm (Fig. 1c, top left). The fluorescence signal was not significantly different in CF-KM4 cells transduced at an inhibitory dose of vector (MOI, 2500 VP/cell): GFP-CFTR was still observed at the plasma membrane (Fig. 1c, top right). The only difference with the MOI 2500 pattern was an increase in fluorescent inclusions within the cytoplasm (Fig. 1c, top right), which again could hardly account for the drastic inhibition of exogenous CFTR activity in cells transduced with high Ad5GFP-CFTR input. This suggested that the inhibitory effect was not due to a decrease in CFTR protein synthesis or some alteration of its cellular trafficking, but rather to a functional mechanism.
Role of viral genome, capsid, and transgene products in the negative interference of Ad5 vector with endogenous CFTR
We then analyzed the influence of Ad5GFP-CFTR vector on the function of endogenous Cl− channels in CFTR-positive tracheal glandular cells (MM-39), the counterpart of CF-KM4 cells. We found a negative effect on endogenous CFTR function in MM-39 cells at high vector doses (2500 VP/cell; Fig. 2a), similar to that observed on exogenous CFTR transferred to CF-KM4 cells. To rule out any possible interference between endogenous and exogenous CFTR molecules in Ad5GFP-CFTR transduced cells, MM-39 cells were transduced with control vector Ad5GFP. A similar profile of endogenous CFTR inhibition was observed at high MOI (2500 VP/cell; Fig. 2b), implying that this negative effect was independent of the CFTR protein delivered by the Ad5 vector. Furthermore, empty Ad5 particles devoid of viral DNA, on incubation with MM-39 cells, exerted a negative effect on CFTR similar to that provoked by Ad5 virions with full genome content, and at similar viral inputs (Fig. 2c). This strongly suggested that the negative effect of Ad5 vector on CFTR was due to the intrinsic nature of the incoming virions, and not to newly synthesized viral proteins or transgene product, or some basal replication of the vector.

Negative effect of Ad5 virions and empty viral particles on the endogenous CFTR Cl− channel at high particle doses. CFTR-positive MM-39 cells were infected with various viral inocula and at increasing MOIs, as indicated on the x axis. Shown are histogram representations of the iodide efflux in response to forskolin plus genistein (10 and 30 μM, respectively) in cells 48 hr after infection with (
Identification of Ad5 capsid protein(s) responsible for negative interference with CFTR
The next issue was to determine whether the negative effect on CFTR was associated with the whole capsid, or carried by an individual viral capsomer (Fig. 2d). With this aim in mind, Ad5 virions were incubated with 0.5% deoxycholate (DOC) at 56°C for 90 sec, a mild treatment that was sufficient to disrupt the adenoviral particle into its internal core (viral DNA associated with core proteins V and VII), apex components (penton capsomer, peripentonal hexons, and protein IIIa), and groups of nine hexons associated with hexon-associated proteins VI, VIII, and IX (Boulanger et al., 1977, 1979). DOC-treated virus showed the same inhibitory effect on Cl− channel function as untreated virions and empty capsids (Fig. 3a; compare with Fig. 2a). This demonstrated that the integrity of the capsid was not required for the negative interference of Ad5 with CFTR, and that free capsid protein(s) could reproduce the negative effect of the whole virus.
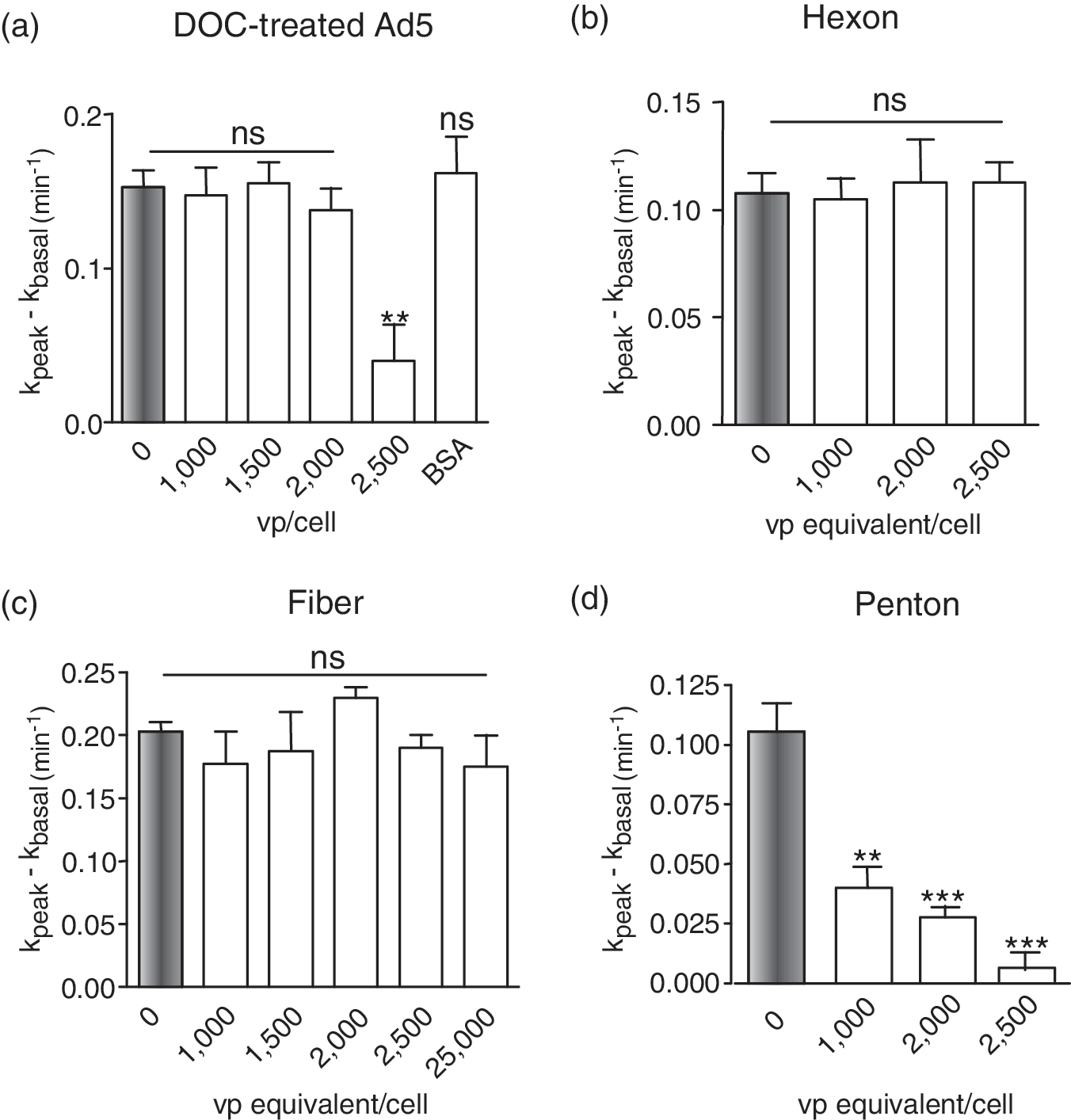
Effect of disrupted Ad5 capsids and free capsomers on the endogenous CFTR Cl− channel. CFTR-positive MM-39 cells were incubated with (
To identify the capsid protein(s) involved, the three major capsid proteins of Ad5 were assayed separately for their effect on endogenous CFTR channels of MM-39 cells. Hexon, penton (penton base and fiber), and fiber proteins (refer to Fig. 2d) were purified from the pool of viral proteins recovered from Ad5-infected cell lysates (Boulanger and Puvion, 1973; Molinier-Frenkel et al., 2002; Franqueville et al., 2008). Each capsid protein was incubated with MM-39 cells at increasing doses, equivalent to 0, 1000, 1500, 2000, 2500, and 25,000 VP/cell. The amount of each protein added was calculated to correspond to its capsid content at each virus dose used, and the protein inputs were therefore expressed as viral particle-equivalents per cell (VP-equivalents/cell).
Hexon and fiber proteins did not exhibit any detectable effect on CFTR up to protein input of 2500 VP-equivalents/cell (Fig. 3b and c). With penton, however, a significant inhibitory effect was detected at 1000 VP-equivalents/cell, and an almost complete inhibition was observed at 2500 VP-equivalents/cell (Fig. 3d). The fact that the penton (base and fiber), but not the free fiber protein, reproduced the negative effect on CFTR indicated that the CFTR-inhibitory function was associated with the penton base domain, and not the fiber.
Involvement of penton base–integrin-mediated endocytic pathway in Ad5-mediated CFTR inhibition
The penton base is a homopentameric protein that displays five RGD tripeptide motifs. These motifs are involved in the endocytosis and cellular internalization pathway of Ad5 virions, via their interaction with αv-integrins (Wickham et al., 1993, 1994, 1995). To assess the role of the penton base and RGD integrin-dependent endocytic pathway in the Ad5-mediated negative effect on CFTR, we transduced MM-39 cells with Ad5PbEGDGFP, an Ad5 vector mutated in the penton base RGD motifs (RGD-to-EGD substitution [Henning et al., 2005; Waszak et al., 2007]). Ad5PbEGDGFP, which bypasses the integrin pathway for infection, shows a slight delay in its growth curve, but has similar productivity and infectivity compared with the parental vector Ad5GFP (Henning et al., 2005; Granio et al., 2009). Ad5PbEGDGFP transduced MM-39 cells and expressed GFP at the same levels as Ad5GFP 48–72 hr posttransduction (data not shown). However, Ad5PbEGDGFP showed no negative effect on Cl− channel activity at MOI up to 2500 VP/cell (Fig. 4a). This result suggested that the negative interference of Ad5 with CFTR involved the penton base RGD motifs, and likely the cellular internalization of virions via the RGD-dependent integrin pathway.
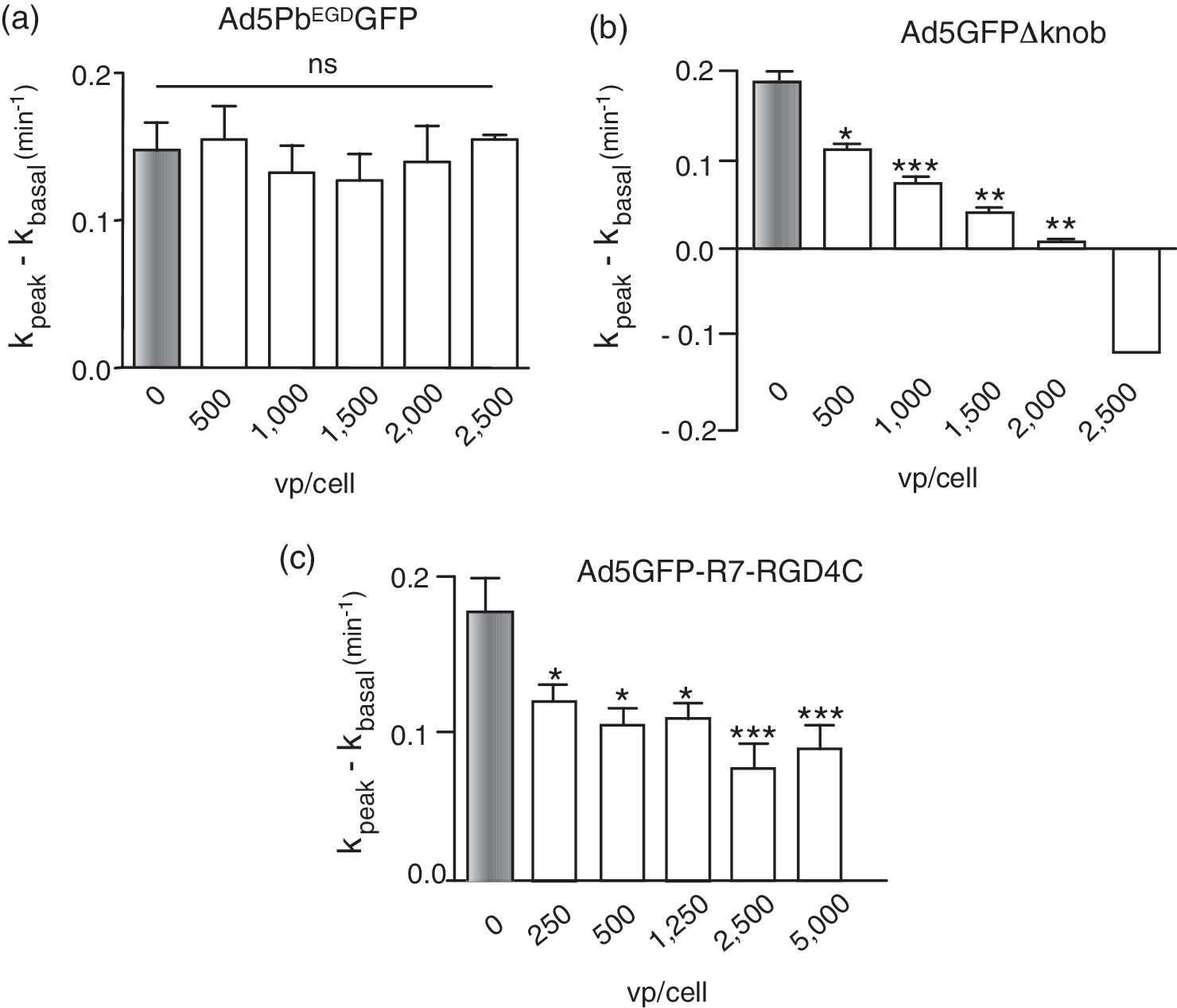
Effect of Ad5GFP vector mutants on the endogenous CFTR Cl− channel. CFTR-positive MM-39 cells were infected with (
This was further supported by the phenotype of two other Ad5 fiber mutants, Ad5GFPΔknob and Ad5GFP-R7-RGD4C. Ad5GFPΔknob carries short fibers that are deleted of the knob and most of the shaft domain, and its penton base capsomers are more accessible to cell surface ligands, compared with wild-type virus (Magnusson et al., 2001; Henning et al., 2006). Ad5GFP-R7-RGD4C carries cyclic, disulfide-constrained RGD motifs at the C terminus of short-shafted fibers. Both Ad5GFP-R7-RGD4C and Ad5GFPΔknob showed a net inhibitory effect on CFTR activity (Fig. 4b and c), confirming the involvement of the penton base RGD motifs, and the absence of a direct role of the fiber knob domain, in this negative effect.
It has been shown that recombinant penton base protein binds to the surface of epithelial cells, is endocytosed and internalized, and follows the intracellular trafficking pathway of adenovirions to reach the nucleus (Hong et al., 1999). It could therefore be hypothesized that, at high Ad5 doses, or high penton protein concentrations, CFTR protein would be coendocytosed with virion or penton base protein, a pathway that involves RGD-dependent integrins. In such a scenario, coendocytosis and cointernalization of virions (or penton base capsomers) with CFTR molecules resident of the plasma membrane of CFTR-positive cells would decrease the number of Cl− channels available at the cell surface, or/and alter the recycling of endogenous CFTR molecules to the plasma membrane. In CFTR-negative cells, cellular uptake of a bulk of Ad5 particles (or penton base capsomers) and saturation of the endosomal compartment could negatively affect the cellular trafficking and membrane addressing of newly synthesized, exogenous CFTR molecules expressed by the adenoviral vector.
To address this issue, MM-39 cells were transduced with Ad5GFP-CFTR at 1250 VP/cell, an MOI that induced a small but significant increase in the activity of the endogenous Cl− channel (about 30%; Granio et al., 2007). At 72 hr after transduction, we considered that the exogenous GFP-CFTR molecules were endogenized, as they incorporated into the pool of endogenous CFTR molecules and followed their metabolic pathway. Transduced MM-39 cells were then incubated with increasing doses of empty Ad5 capsids, and examined by confocal fluorescence microscopy and live-cell imaging. No increased endocytosis of GFP-CFTR could be detected in capsid-treated cells, compared with control, untreated cells (data not shown). This further supported the hypothesis that the negative effect of Ad5 vector at high doses did not result from altered biosynthesis and/or cellular trafficking of the CFTR protein, but rather from an indirect mechanism of blockage of CFTR activity.
Absence of penton base-associated negative interference with CFTR by Ad5 vector pseudotyped with serotype 35 fiber
Because the previous results suggested that the negative interference with CFTR function depended on the RGD-integrin endocytic pathway, we analyzed the CFTR activity of cells transduced with an Ad5-based modified vector that has been reported to enter cells via an alternative pathway. The chimeric Ad5F35-GFP vector carries serotype 35 fibers on an Ad5 capsid, and uses CD46 and heparan sulfate proteoglycans as cellular receptors (Gaggar et al., 2003; Tuve et al., 2008). When tested on MM-39 cells at increasing MOIs, Ad5F35-GFP showed no negative effect on the activity of endogenous CFTR channels, even at MOIs up to 5000 VP/cell (Fig. 5a).
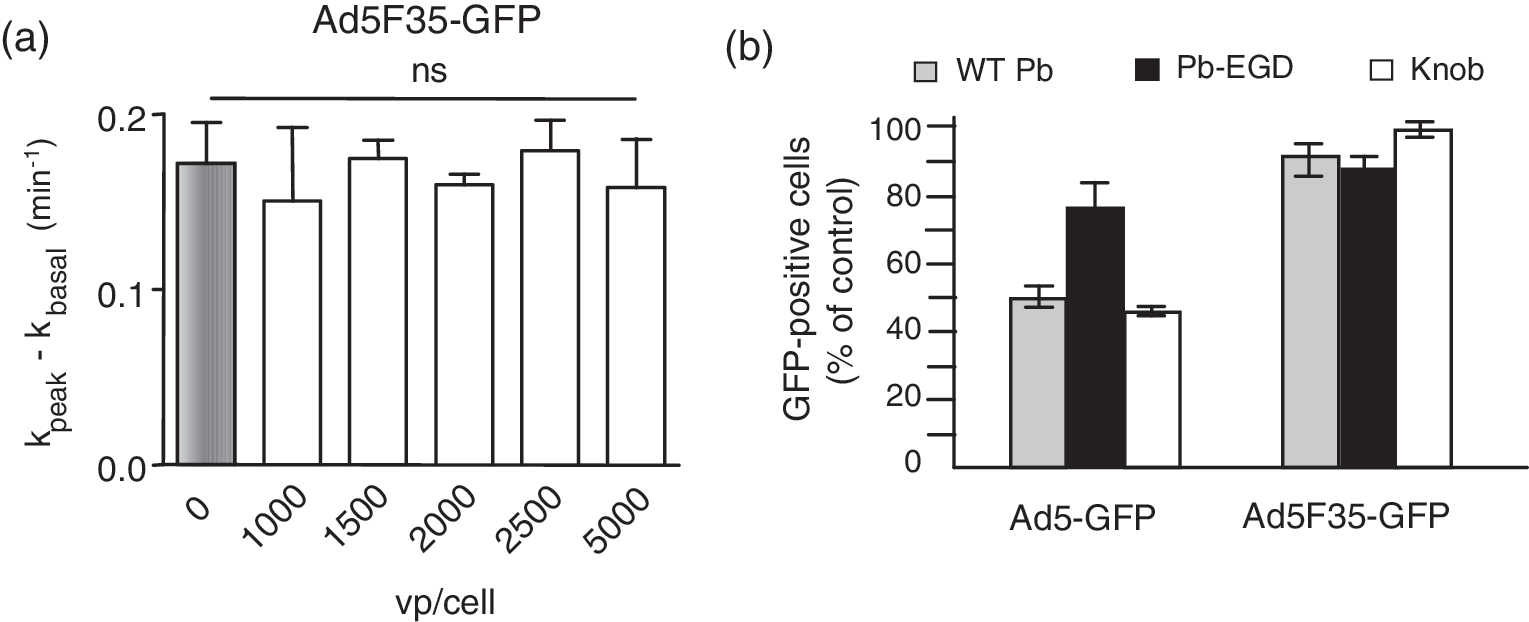
Absence of negative effect of fiber-pseudotyped vector Ad5F35-GFP on the endogenous CFTR Cl− channel, and cell entry pathway. (
To further analyze the mechanism of cellular uptake of Ad5F35-GFP vector in relation to CFTR channel activity, we performed competition assays using recombinant fiber knob and penton base proteins during the phase of cell entry at 37°C, after virus–cell attachment at 4°C (Gaden et al., 2002, 2004). The amount of penton base or knob competitor that blocked transduction by Ad5GFP vector by 50% failed to provoke any detectable effect on Ad5F35-GFP vector (Fig. 5b). Control experiments using the EGD mutant of penton base protein showed no effect on either vector (Fig. 5b). These results suggested that the cell entry pathway of Ad5F35-GFP differed from the penton base-RGD-integrin internalization pathway used by Ad5GFP.
The absence of any negative effect of the Ad5F35-GFP vector on CFTR was not due to a lack of cell transduction, as shown in Fig. 6. The efficiency of cell transduction by chimeric vector Ad5F35-GFP was compared with that of Ad5GFP used at the same MOI in three airway cell lines: MM-39, CF-KM4, and A549. For the three cell lines, and at all vector doses tested, Ad5F35-GFP showed higher transduction efficiency (Fig. 6a) compared with Ad5GFP (Fig. 6b), as assayed by the number of GFP-positive cells and the intensity of the fluorescence signal in flow cytometry (data not shown). This indicated that the cell entry pathway used by Ad5F35-GFP was more efficacious than the one used by Ad5GFP to enter airway cells.
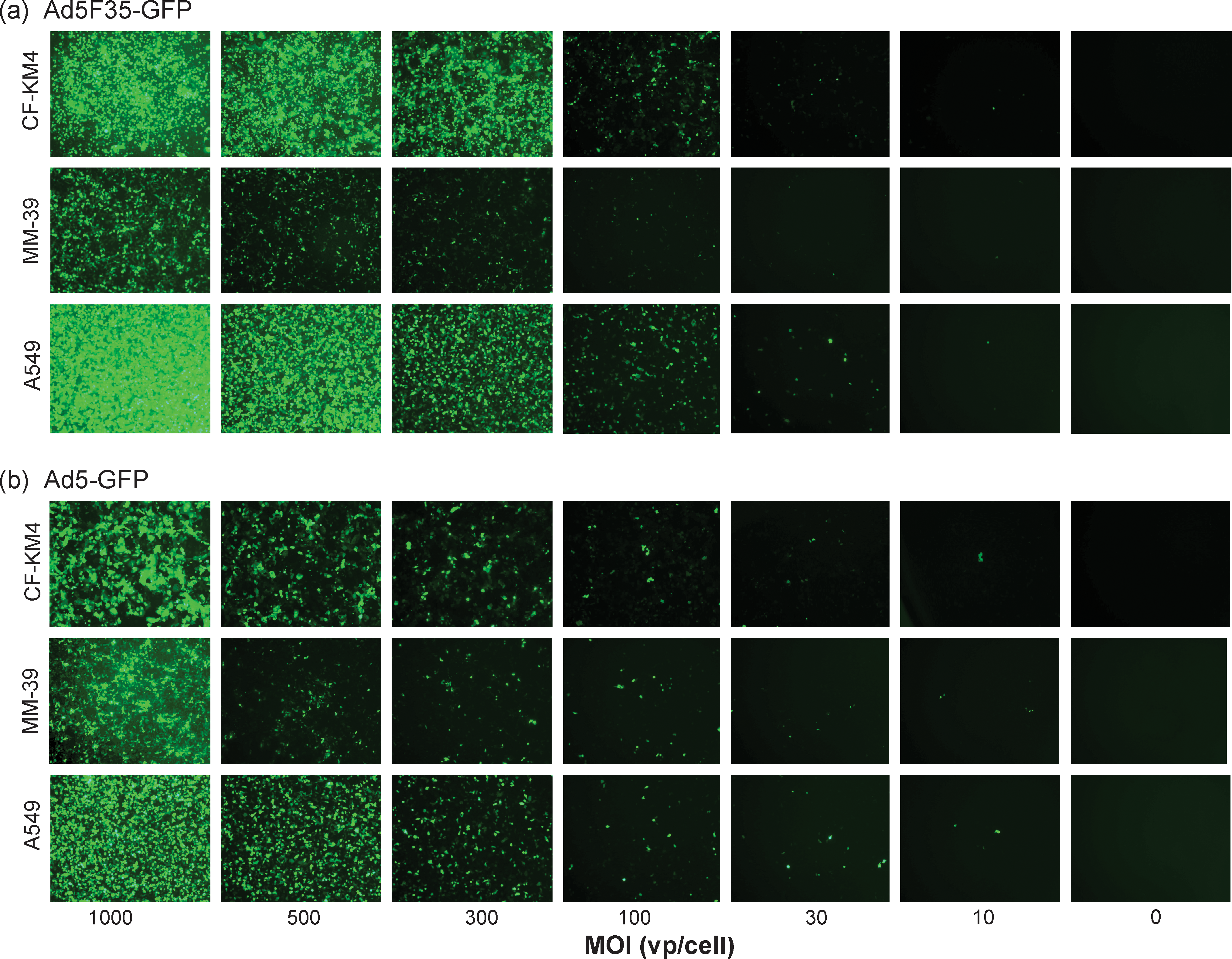
Comparison of cell transduction efficiency between Ad5F35-GFP and Ad5GFP vectors in vitro. Confluent monolayers of CF-KM4 cells (top rows), MM-39 cells (middle rows), and A549 cells (bottom rows) were infected with (
Efficient correction of Cl− channel activity in CFTR-deficient cells transduced with Ad5F35-GFP-CFTR
The capacity of Ad5F35-GFP-CFTR to restore CFTR Cl− channel function was first analyzed in the CFTR-deficient tracheal glandular cell line CF-KM4. A significant correction of Cl− channel activity was detected 48 hr after transduction at an MOI as low as 50 VP/cell (Fig. 7A, panel a), corresponding to an infectious titer of only 2 PFU/cell, as assayed in HEK-293 cells. By comparison, to obtain the same level of correction of Cl− channel function by Ad5GFP-CFTR in CF-KM4 cells, a 25- to 30-fold higher vector input was required, that is, an MOI of 1250–1500 VP/cell (Granio et al., 2007). In Ad5F35-GFP-CFTR-transduced CF-KM4 cells, Cl− channel activity increased in a vector dose-dependent manner starting at 50 VP/cell, with no detectable inhibition even at high MOIs (2000 VP/cell; Fig. 7b). These results demonstrated that the fiber 35-pseudotyped vector Ad5F35-GFP-CFTR was more efficient in gene transduction and Cl− channel deficiency correction than Ad5GFP-CFTR at the same vector dose. More importantly, Ad5F35-GFP-CFTR showed no deleterious effect on the function of exogenous CFTR channel at high vector inputs.
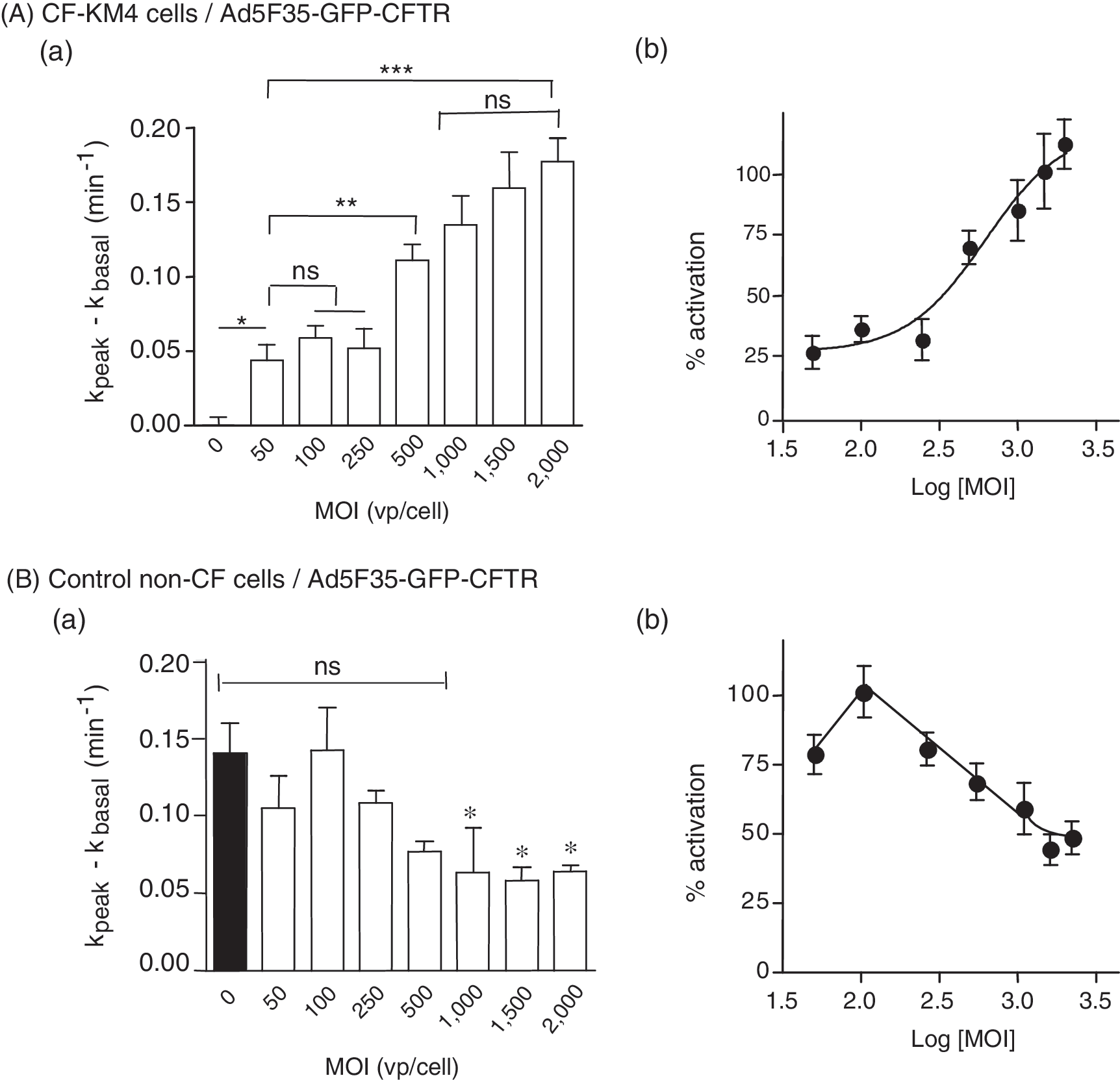
Effect of Ad5F35-GFP-CFTR on Cl− channel activity in human cell lines in vitro. (
A different profile was observed when the control, non-CF cell line MM-39 was transduced with Ad5F35-GFP-CFTR at increasing vector doses. After a slight increase at low vector dose (MOI, 100), the CFTR activity decreased and plateaued at 45–50% of the basal activity at an MOI of 1000 and above (Fig. 7B, panel a and b). Because cell entry by Ad5F35-GFP virions did not produce any detectable CFTR dysfunction (refer to Fig. 5a), this would suggest some degree of interference between cell-regulated endogenous CFTR and upregulated, CMV promoter-driven exogenous CFTR.
Efficient apical transduction of reconstituted human airway epithelia by Ad5F35-GFP-CFTR
Primary CFTR-deficient human airway epithelial (CF-HAE) cells grown as reconstituted airway epithelia were infected with Ad5F35-GFP-CFTR (or control Ad5GFP-CFTR) at an MOI of 2, 20, or 200 VP/cell via the apical surface, and observed by fluorescence microscopy. In Ad5GFP-CFTR-treated samples, no GFP-positive cells were observed at MOIs of 2 or 20 (data not shown). With Ad5F35-GFP-CFTR, some GFP-positive cells were observed at MOI 2, and 8–12% of cells were found to be GFP positive at MOI 20 (data not shown). However, at MOI 200, GFP-positive cells were detected in CF-HAE samples transduced with either vector (Fig. 8A, panels a and b). An estimation of their respective transduction efficiencies was given by GFP-positive cell counts determined 96 hr posttransduction (Fig. 8B): 233.66 ± 12.73 GFP-positive cells per field of view were counted for Ad5F35-GFP-CFTR, versus 19.33 ± 3.62 for Ad5GFP-CFTR (mean ± SE; n = 3), that is, a 10-fold difference in favor of Ad5F35-GFP-CFTR.
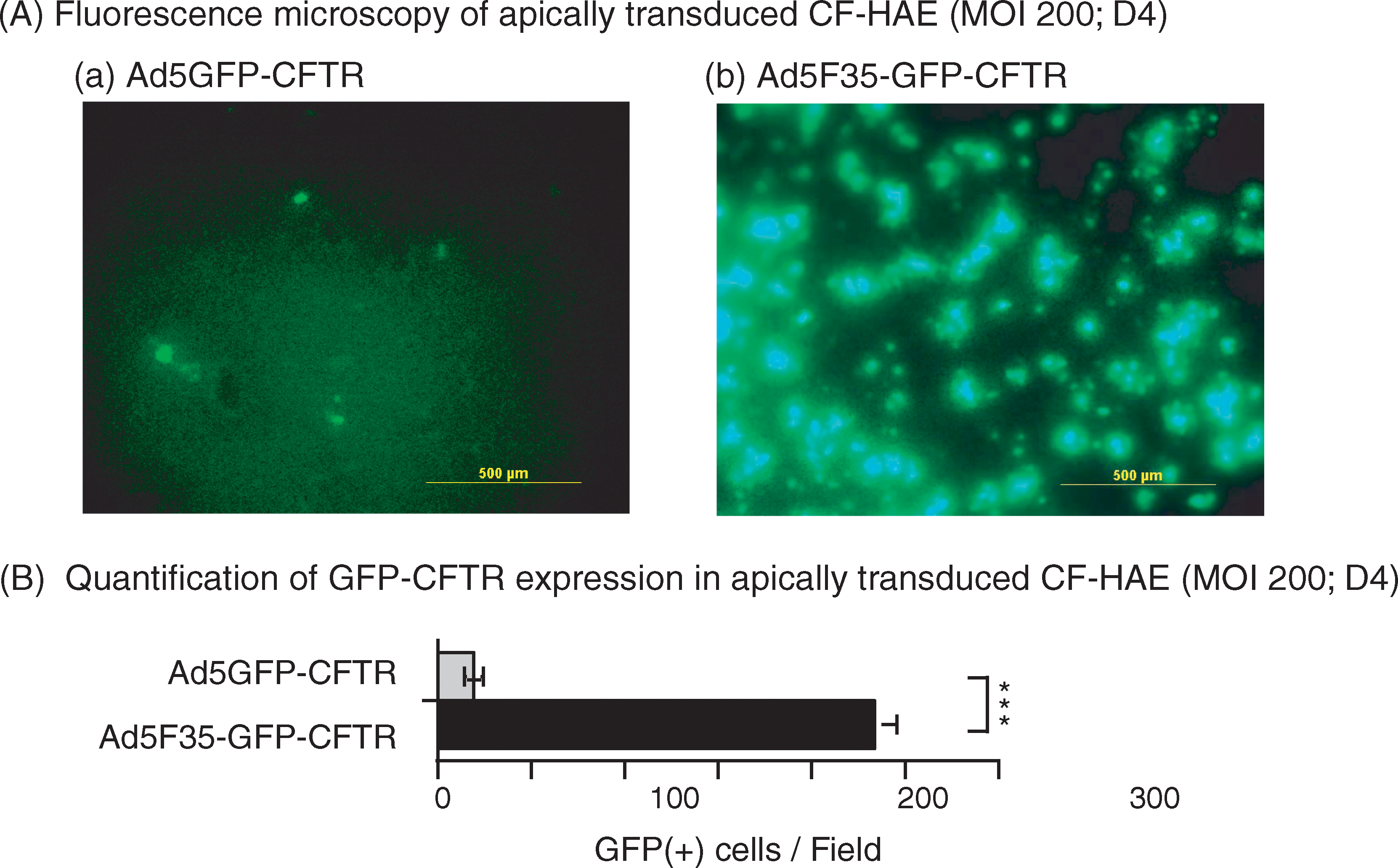
Efficient apical transduction by Ad5F35-GFP-CFTR of ex vivo-reconstituted human airway epithelia (HAE) from patients with CF (CF-HAE). (
We next analyzed by fluorescence confocal microscopy the cellular localization of GFP-CFTR in CF-HAE after apical application of Ad5F35-GFP-CFTR vector at various MOIs (Fig. 9). In particular, it was essential to determine whether GFP-CFTR protein localized at the apical pole of the reconstituted epithelia. We used immunofluorescence staining of ZO-1 as a marker of apical tight junctions (Fig. 9, red signal). At an MOI of 20, the average number of cells expressing GFP-CFTR protein at the apical pole was 13.8 ± 0.42 (mean ± SE; n = 3; Fig. 9A). At an MOI of 200, the proportion of cells displaying GFP-CFTR at the apical pole increased by a factor of more than 3, 46.00 ± 7.13 (mean ± SE; n = 3; Fig. 9B).
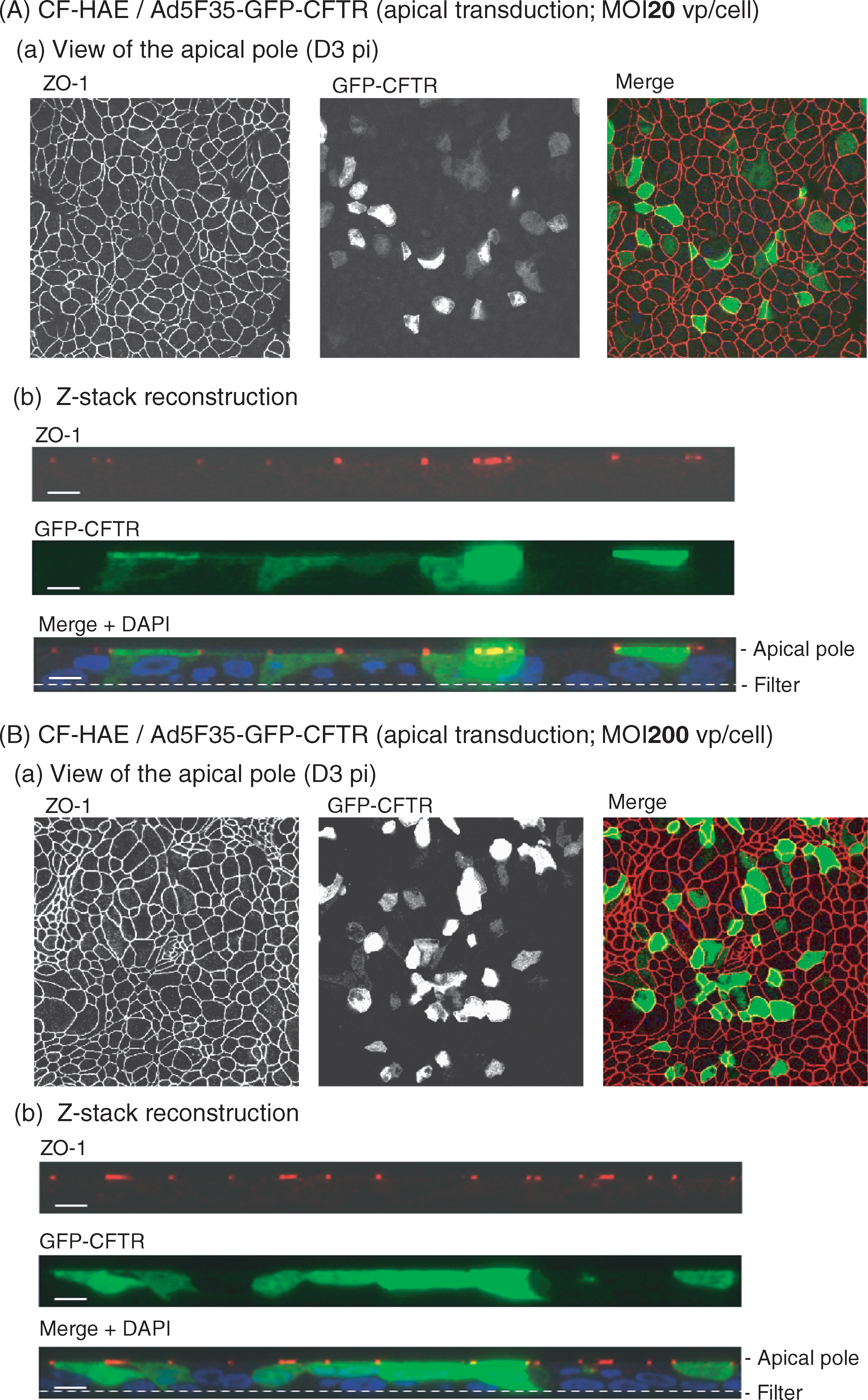
Expression and apical localization of GFP-CFTR fusion protein in CF-HAE apically transduced with Ad5F35-GFP-CFTR. CF-HAE were apically infected with Ad5F35-GFP-CFTR at an MOI of (
Ad5F35-GFP-CFTR-mediated correction of CFTR deficiency in CF-HAE
It was documented that in CF-HAE, more than 5% of the cells must be corrected to see a change in CFTR chloride current (Farmen et al., 2005). More recently, it has been shown that 25% of cells must be transduced with a CFTR-expressing parainfluenza viral vector to restore the normal rate of mucus transport (Zhang et al., 2009). In the present study, we analyzed the bioelectrical properties of Ad5F35-GFP CFTR-transduced CF-HAE samples at an MOI of 200, and at 72 hr after apical infection, in Ussing chambers, as previously described (Ashbourne Excoffon et al., 2003; Ostedgaard et al., 2003; Farmen et al., 2005; Granio et al., 2007). The transepithelial Cl− current was measured under short-circuit conditions (I sc), and the data were compared with those obtained with CF-HAE transduced with control vector Ad5GFP-CFTR administered at the same vector dose (MOI, 200 VP/cell), and similarly inoculated at the apical pole. Ad5F35-GFP-CFTR restored CFTR function in CF-HAE cells, whereas no correction was detected with the same dose of control vector Ad5GFP-CFTR (Fig. 10A). These functional data confirmed the epifluorescence pattern shown previously (refer to Fig. 8A).
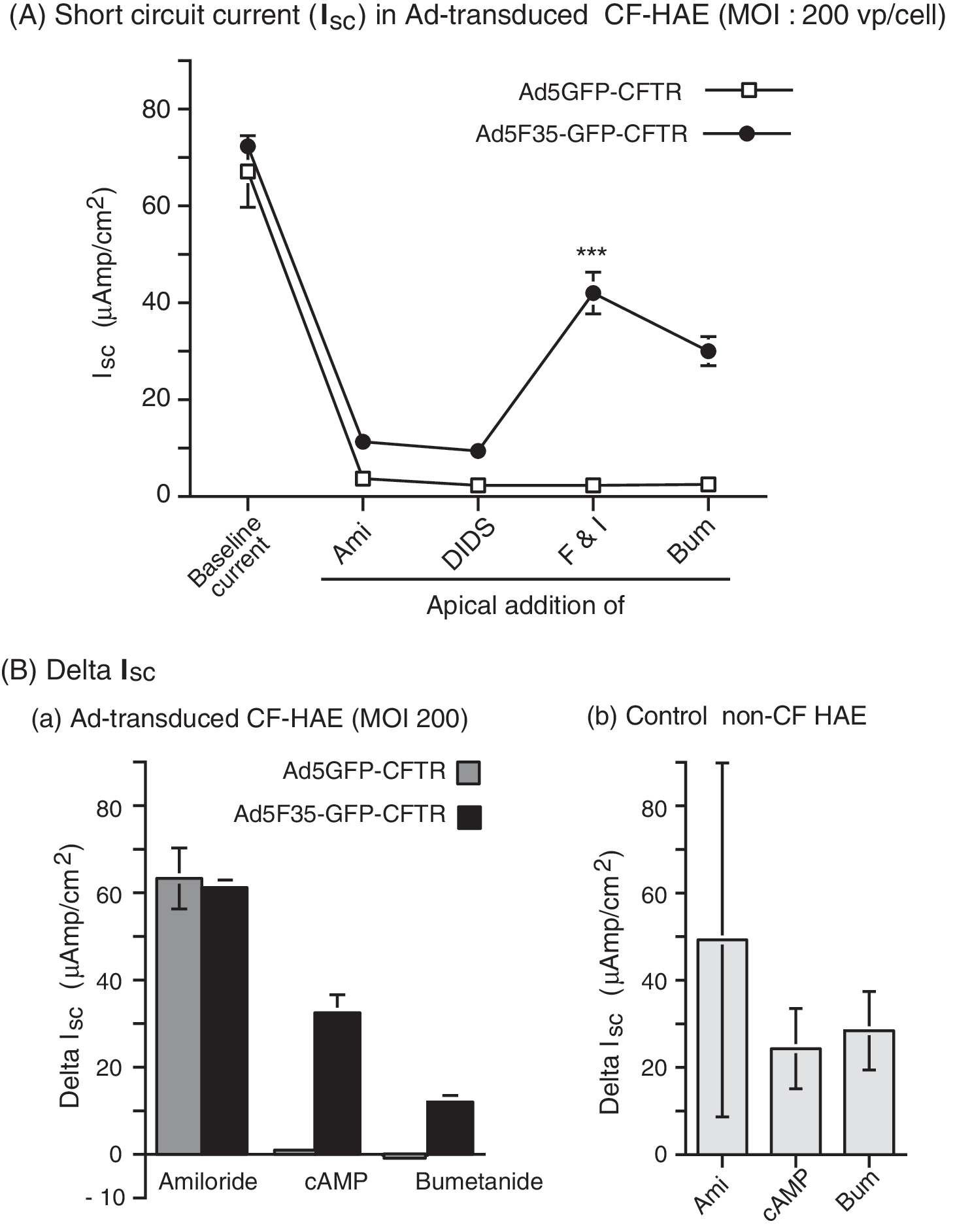
Transepithelial current in CF-HAE after adenovirus-mediated CFTR transfer via the apical pole. Well-differentiated epithelia from patients with CF (CF-HAE) were apically infected with Ad5GFP-CFTR (open symbols) or Ad5F35-GFP-CFTR (solid symbols), at an MOI of 200 VP/cell. Three days after adenoviral transduction, CF-HAE were analyzed in Ussing chambers for transepithelial Cl− current, measured under short-circuit conditions (I
sc). (
When graphically plotted, the ΔI sc values obtained for CF-HAE transduced with Ad5F35-GFP-CFTR versus Ad5GFP-CFTR indicated that Ad5F35-GFP-CFTR was 30 times more efficacious than Ad5GFP-CFTR in correcting the CFTR deficiency via the apical pole, and at a relatively low vector dose (200 VP/cell; Fig. 10B, panel a). The level of CFTR activity in CF-HAE corrected by Ad5F35-GFP-CFTR was in the range of experimental values measured as the transepithelial Cl− current in control, non-CF epithelia (Fig. 10B, panel b). These results demonstrated that the Ad5F35-GFP-CFTR vector was able to efficiently transduce CF-HAE via the apical surface and correct the chloride transport defect. However, Ad5GFP-CFTR was totally inefficient at the same vector dose (200 VP/cell) when inoculated at the apical surface, as previously observed (Walters et al., 1999; Zabner et al., 1999; Granio et al., 2007).
Duration of GFP-CFTR expression in apically transduced HAE
A comparative time course analysis of GFP-CFTR expression was performed in non-CF HAE transduced apically with Ad5F35-GFP-CFTR or Ad5GFP-CFTR, at two different vector doses (MOIs of 200 and 600 VP/cell). At both MOIs the proportion of GFP-positive cells in Ad5F35-GFP-CFTR-transduced HAE increased from day 3 to day 6 posttransduction, followed by a slight decrease, and remained stable for at least 5 weeks. With Ad5GFP-CFTR, however, the number of GFP-positive cells decreased progressively with time (Fig. 11a and b). The difference between the two vectors was even more pronounced at an MOI of 600 (Fig. 11b), and confirmed the efficacy of Ad5F35-GFP-CFTR in terms of long-term expression of CFTR in HAE after apical application of the vector.

Duration of GFP-CFTR expression in HAE after adenovirus-mediated gene transfer via the apical pole. Non-CF HAE were apically transduced with Ad5F35-GFP-CFTR (solid symbols) or Ad5GFP-CFTR (open symbols), at MOIs of (
Discussion
In a previous study, we showed that when CFTR-deficient human tracheal glandular cells (CF-KM4) were transduced with an Ad5GFP-CFTR vector, correction of the chloride channel deficiency was partial, and that increasing the vector dose (>2000 VP/cell, corresponding to 60–80 PFU/cell) was detrimental to CFTR function (Granio et al., 2007). Because it was reported that the overexpression of CFTR protein could lead to its altered trafficking and ectopic cellular localization (Farmen et al., 2005), we quantitatively analyzed the level of GFP-CFTR expression and stability. We found no increase in the cellular content of GFP-CFTR at high vector doses, but instead a slight decrease at late times after transduction, a discrete effect that could hardly account for the drastic inhibition of CFTR channel function. Fluorescence microscopy of cells transduced at low and high doses of Ad5GFP-CFTR showed no difference in membrane localization of GFP-CFTR in both cell samples. The minor differences observed in the cellular distribution of GFP-CFTR could not explain the negative effect of high vector inputs on CFTR function, and suggested a qualitative and functional effect rather than a quantitative alteration or mistrafficking of the CFTR.
Unexpectedly, we observed the same negative effect on the channel activity of endogenous CFTR in MM-39 cells, the CFTR-positive counterpart of CF-KM4 cells (Gaden et al., 2002, 2004), after transduction with Ad5GFP-CFTR and Ad5GFP at high vector doses. The fact that the same effect occurred with control vector Ad5GFP indicated that it was not due to some interference between endogenous and exogenous CFTR molecules. The negative effect on CFTR activity was reproduced by genome-devoid, empty capsids of Ad5, implying that the CFTR inhibition was not due to the genome of incoming virions, or to a basal level of viral replication within transduced cells, but rather to the capsid itself. We therefore analyzed the effect of the three major capsid proteins on CFTR function, assayed individually as hexon, fiber, and penton (base plus fiber). We found that the determinants of the negative interference with CFTR were carried by the penton base domain of the apical capsomer penton, and not by the hexon or fiber domain.
We then tested three penton fiber-modified vectors, Ad5PbEGDGFP, Ad5GFPΔknob, and Ad5GFP-R7-RGD4C, in MM-39 cells at increasing MOIs. Both Ad5GFPΔknob and Ad5GFP-R7-RGD4C showed a strong negative effect on endogenous CFTR, whereas no effect was observed with Ad5PbEGDGFP. This confirmed that the negative interference with CFTR was not associated with the fiber, but with the penton base protein and more specifically its RGD motifs, and most likely involved the RGD-integrin-mediated endocytic pathway. Analysis of GFP-CFTR-expressing cells by confocal microscopy and live-cell imaging excluded Ad5-induced endocytosis of GFP-CFTR molecules. Taken together, our results suggested that the Ad5 penton base-mediated negative interference with CFTR did not result from a direct effect on the biosynthesis or cellular trafficking of the CFTR protein, but rather from an indirect mechanism of blockage of CFTR function.
The RGD motifs of penton base capsomers have been identified as major determinants of the early cytopathic effect induced by subgroup C adenoviruses, such as Ad2 or Ad5, because of their interaction with cellular integrins and their role in the cell-detaching effect, as well as their function in the integrin-mediated endocytic pathway of the virus and its vesicular escape (Seth et al., 1984, 1985; Karayan et al., 1994, 1997; Seth, 1994). Binding of penton base RGD motifs to αV integrins has been shown to induce several integrin signaling pathways such as the phosphoinositide-3-kinase (PI3K) pathway (Philpott et al., 2004), and to induce RANTES (regulated on activation, normal T cell expressed and secreted) expression implicated in the acute inflammatory process induced by adenoviral vectors (Bowen et al., 2002). The Toll-like receptor (TLR) pathway has also been found to be activated in adenovirus-infected epithelial cells (Hartman et al., 2007; Hensley and Amalfitano, 2007), involving TLR4 (Thorne et al., 1999) or TLR9 molecules (Cerullo et al., 2007; Iacobelli-Martinez and Nemerow, 2007). In epithelium-derived and endothelial cells, the induction of host inflammatory genes was reduced with CAR-ablated and RGD deletion-modified adenoviral vectors (Liu et al., 2003).
The present study identified a novel aspect of the penton base RGD-mediated cytopathic effect, which consisted of negative interference by the incoming Ad5 particles with CFTR channel function. This effect is part of the global cytotoxicity of the adenovirus, and must be taken into consideration in gene therapy protocols using Ad5-derived vectors for the correction of a disease. However, RGD-mediated CFTR inhibition would not be an issue when Ad5 vectors are used in tissue destruction strategies, for example, in the case of oncolytic adenoviruses.
With the aim of designing a more efficient vector devoid of CFTR inhibition, the fibers of the Ad5GFP-CFTR vector were swapped for serotype 35 fibers, generating the chimeric vector Ad5F35-GFP-CFTR. We found that the Ad5F35-GFP vector did not interfere with CFTR function and showed increased efficiency in transducing MM-39 cells, CF-KM4 cells, and most of the epithelial cell lines tested, as compared with Ad5GFP. In CFTR-deficient CF-KM4 cells, Ad5F35-GFP-CFTR restored CFTR channel function at significantly lower MOIs, compared with Ad5GFP-CFTR: detectable CFTR activity was already observed at an MOI of 50 VP/cell, with a maximum at an MOI of 1000 VP/cell. By contrast, in cells transduced by Ad5GFP-CFTR, CFTR activity was detectable only at MOIs ≥ 1250–1500 VP/cell (equivalent to 50–60 PFU/cell; Granio et al., 2007).
Competition experiments with penton base and fiber proteins indicated that the fiber 35-pseudotyped vector Ad5F35-GFP entered cells via a route that differed from that of Ad5, and did not depend on penton base RGD–integrin interaction. It has been reported that an Ad5-based vector bearing chimeric fibers composed of the Ad5 shaft and the Ad35 knob domain required integrins for internalization (Shayakhmetov et al., 2005). This did not seem to be the case for our Ad5F35 chimera, which carried the entire fiber of serotype 35 bound to serotype 5 penton base, and bypassed the penton base–integrin internalization pathway. One study has shown that Ad35 binds cellular heparan sulfate proteoglycans via its fiber shaft domain, and the data suggested that CD46 and heparan sulfate proteoglycans could play the role of receptor and/or coreceptor for Ad35 endocytosis (Tuve et al., 2008).
The superiority of the Ad5F35-GFP vector was confirmed by transduction assays using CF HAE, primary epithelia from CF patients reconstituted ex vivo: Ad5F35-GFP-CFTR restored CFTR function at significantly lower vector doses, compared with Ad5GFP-CFTR. More importantly, our chimeric vector Ad5F35-GFP-CFTR was capable of transducing CF-HAE cells via the apical pole, a cell entry pathway that is a critical parameter for gene transfer to airway tissues in vivo and ex vivo. In addition, GFP-CFTR expression remained stable for at least 5 weeks in HAE transduced with Ad5F35-GFP-CFTR. Chimeric Ad5F35 is therefore a more efficient vector compared with Ad5, in terms of CFTR gene delivery and CFTR protein expression and activity, and lacks any adverse effect on CFTR function. It has been reported that no benefit in terms of survival was observed after in utero administration of conventional vector Ad5-CFTR to fetal CFTR-knockout mice (Buckley et al., 2008; Davies et al., 2008). It would therefore be interesting to test our Ad5F35-CFTR vector in a similar in vivo protocol.
The relevance of the Ad5 negative effect on CFTR on differentiated human airway epithelia remains unclear because of the complexity of this cell model system. Previous studies have demonstrated that correction of a minimum of 5% of epithelial cells is required in order to observe a change in CFTR current (Farmen et al., 2005), a level hardly achievable by the Ad5 vector when inoculated at the apical surface. Interestingly, in this same study basolateral transduction of non-CF epithelia by Ad5-βGal at an MOI of 50,000 VP/cell did not alter endogenous CFTR activity, suggesting that either this phenotype is not present in this cell type or that infection from the basolateral surface differs from the pathway followed in other cell types and potentially from the apical surface. Indeed, several viruses have distinct trafficking pathways when infected from the apical surface in comparison with the basolateral surface (Duan et al., 2000; Jia et al., 2006; Vermeer et al., 2007; Excoffon et al., 2008). Elucidation of the molecular mechanism by which viral structural components, such as the Ad5 penton base, negatively interfere with CFTR function might help better our understanding of the various facets of adenovirus pathogenesis, and in turn might lead to subsequent improvement of adenoviral vectors.
Footnotes
Acknowledgments
This work was supported by the French Cystic Fibrosis Foundation (Vaincre la Mucoviscidose, VLM contracts TG0702 and TG0801), the Centre National Recherche Scientifique (CNRS FRE-3011), the University of Lyon I, and the Hospices Civils de Lyon (HCL). O.G. was financially supported by a postdoctoral fellowship from VLM. S.S.H. is an INSERM scientist and the recipient of a Contrat d'Interface HCL-INSERM. The authors are grateful to Fatiha Najioullah for help with RT-PCR analysis, to Pascal Fender (IBS, Grenoble) for help with live-cell fluorescence microscopy, to Nick Gansemer and Sylvie Fiorini for technical assistance, and to Cathy Berthet for efficient secretarial aid.
Author Disclosure Statement
The authors have no competing financial interests.