
Abstract
Lentiviral vectors are an important tool for gene transfer research and gene therapy purposes. However, the low stability of these vectors affects their production, storage, and efficacy in preclinical and clinical settings. In the present work the mechanism underlying the thermosensitivity of lentiviral vectors was evaluated. For lentiviral vectors pseudotyped with amphotropic and RDpro envelopes, the capacity to perform reverse transcription was lost rapidly at 37°C, in high correlation with the loss of infectivity. The vector with RDpro envelope presented a higher level of stability than that with amphotropic envelope for both the reverse transcription process and viral infectivity. Reverse transcriptase enzyme inactivation and viral template RNA degradation were not implicated in the loss of the viral capacity to perform reverse transcription. Furthermore, early entry steps in the infection process do not determine the rate of viral inactivation, as the amount of viral RNA and p24 protein entering the cells decreased slowly for both vectors. Taken together, it can be concluded that the reverse transcription process is thermolabile and thus determines the rate of lentiviral inactivation. Strategies to stabilize the reverse transcription process should be pursued to improve the applicability of lentiviral vectors in gene therapy.
Introduction
One of the major challenges of these vectors is their production, because the development of stable producer cell lines has been a major hurdle, mainly due to the intrinsic cytotoxicity of constitutive expression of some lentiviral proteins (Breckpot et al., 2007). Although producer cell lines have already been developed (Klages et al., 2000; Ikeda et al., 2003; Broussau et al., 2008), lentiviral vectors are still mostly produced transiently by transfection of 293T cells with three or four coding plasmids (Zhang et al., 2001; Farley et al., 2007).
The production of lentiviral vectors is affected both by low titers and by the low stability of vector infectivity. The low stability of these vectors at 37°C is a critical issue with respect to production, storage, and the efficacy of viral preparations in clinical studies; nevertheless, the viral stability issue has been poorly addressed (Higashikawa and Chang, 2001; Strang et al., 2004). Several viral components may be susceptible to degradation/inactivation affecting viral infectivity namely, genomic viral RNA, capsid proteins, envelope proteins, and the enzymes reverse transcriptase (RT) and integrase. Our laboratory showed that a key inactivation mechanism of MLV-based gammaretroviral vectors at 37°C is the loss of viral capacity to perform reverse transcription (Carmo et al., 2008), raising the possibility of a similar inactivation mechanism affecting lentiviral vectors.
The capacity to produce lentiviral vectors pseudotyped with various envelope proteins is critical to enable gene targeting to specific tissues and cells in clinical applications (Verhoeyen and Cosset, 2004; Cronin et al., 2005). Vesicular stomatitis virus glycoprotein G (VSV-G) envelope (VSV-G Env) is often used to pseudotype lentiviral vectors. However, to ensure reproducible vector quality among productions, a stable producer cell line was chosen in this work; given its high cytotoxicity the VSV-G pseudotype cannot be used in stable producer cell lines (Cronin et al., 2005; Breckpot et al., 2007). Furthermore, VSV-G pseudotypes are inactivated by human serum complement, another drawback of this envelope protein (Depolo et al., 2000; Strang et al., 2004). Hence, two different proteins, the amphotropic and RDpro envelopes (ampho Env and RDpro Env), were selected as pseudotypes in this work; both are resistant to human serum complement, infect clinical relevant cells, and can be expressed in stable producer cell lines (Stitz et al., 2000; Sandrin et al., 2002; Strang et al., 2004; Cronin et al., 2005). In addition, it is known that vectors pseudotyped with the RD114 envelope are more thermostable than vectors pseudotyped with amphotropic envelope (Cosset et al., 1995). The ampho Env (derived from murine leukemia virus) efficiently pseudotypes lentiviral vectors whereas the RD114 Env (derived from feline endogenous virus) has a low capacity to pseudotype lentiviral vectors (Sandrin et al., 2002). Consequently, RD114 Env was engineered to be used in lentiviral vectors, by substitution of the cytoplasmic tail with that of MLV Env (Sandrin et al., 2004). Furthermore, the R peptide cleavage site sequence was replaced with that of a matrix–capsid cleavage site in HIV Gag to create RDpro (Strang et al., 2004). This study evaluates the thermosensitivity of the reverse transcription process in lentiviral vectors pseudotyped with various envelope proteins and its correlation to the rapid vector inactivation at physiological temperatures. The data presented herein indicate that the envelope protein affects the thermostability of the viral reverse transcription complex and thus vector infectivity.
Materials and Methods
Lentiviral vector production and purification
Enhanced green fluorescent protein (eGFP)-encoding lentiviral vectors pseudotyped with amphotropic envelope and lentiviral vectors pseudotyped with RDpro envelope were obtained from supernatant of the STAR-A cell line (ECACC no. 04072118; European Collection of Cell Cultures [ECACC], Salisbury, UK) and of STAR RDpro cell line (ECACC no. 04072117), respectively, cell lines developed at the University College London (Ikeda et al., 2003). Both cells lines were cotransfected with pSELECT-puro-mcs (InvivoGen, Toulouse, France) and pSIN-CSGW (kindly provided by A.J. Thrasher, University College London, London, UK) at a ratio of 1:20 and selected with puromycin (3 μg ml−1). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, Steinheim, Germany) supplemented with glucose (4.5 g liter−1) (Merck, Darmstadt, Germany), 2 mM glutamine (GIBCO, Paisley, UK), and 10% (v/v) fetal bovine serum (FBS; GIBCO). After 3 days of culture the medium was replaced, lentiviral vectors were produced during the following 24 hr, and the supernatant was filtered with 0.45-μm filters. Lentiviral vectors without envelope protein and with VSV-G Env were produced by transient transfection of 293T cells. For the production of vectors without Env cells were cotransfected using polyethylenimine (PEI; Sigma-Aldrich) with packaging plasmid p8.91 (Zufferey et al., 1997) and transgene plasmid pSIN-CSGW, and for the production of vectors with VSV-G Env the envelope plasmid pMD2.G (Zufferey et al., 1997) was further used. Twenty-four hours after transfection the medium was replaced, lentiviral vectors were produced during the following 24 hr, and the supernatant was filtered with 0.45-μm filters. All the viral supernatants were concentrated by ultracentrifugation in a Beckman Optima XL-100 centrifuge (Beckman Coulter, Fullerton, CA): 70,000 × g for 1.5 hr at 4°C, using a Beckman 45Ti rotor (Reiser, 2000; Hanawa et al., 2002). The pelleted virus was resuspended in storage buffer (10 mM Tris [Calbiochem, Darmstadt, Germany] [pH 7.2], 2 mM MgCl2 [Merck], and 0.01% Tween 80 [Merck]) and purified by centrifugation on a 20% (w/v) sucrose (Fluka, Steinheim, Germany) solution at 100,000 × g for 2 hr at 4°C, using a Beckman 90Ti rotor (Mok et al., 2007). The final viral pellet was resuspended in storage buffer. The total yield of the concentration and purification steps was 15% for vectors pseudotyped with amphotropic Env and 20% for vectors pseudotyped with RDpro Env. The titer of the final viral preparation was 7 × 106 infectious particles (IP) ml−1 for vectors with amphotropic Env and 4 × 106 IP ml−1 for vectors with RDpro Env.
Inactivation studies
After resuspension of the viral pellet in storage buffer, the viral preparations were incubated at 37°C and samples were removed, before incubation (zero time) and 0.5, 1, 1.5, 2, 4, 6, 8, and 10 hr postincubation. These samples were aliquoted for each test and stored at −85°C. Preincubated samples were thawed and tested for residual infectivity, endogenous and intracellular reverse transcription activity, reverse transcriptase activity, RNA degradation, RNA entry, and p24 entry. The same set of preincubated viral samples was used to perform all tests. Activities in viral samples, without preincubation (zero time), were taken as 100%.
Quantitation of infectious lentiviral vectors
To determine lentiviral infectivity, 293T target cells were seeded in 24-well, flat-bottomed plates (Nunc, Roskilde, Denmark) at a density of 5 × 104 cells per well and incubated for 24 hr. Infections were carried out by replacing the medium with 150 μl of dilutions of viral samples in DMEM containing Polybrene (8 μg ml−1; Sigma-Aldrich), followed by incubation at 37°C for 4 hr. After adsorption, viral solutions were removed and 500 μl of fresh medium was added. At 48 hr postinfection the cells were trypsinized and resuspended, and the percentage of cells expressing GFP was determined by flow cytometry with a CYFlow space flow cytometer (Partec, Münster, Germany). Three dilution sets were performed for each sample tested.
Endogenous reverse transcription
Endogenous DNA synthesis reactions were performed as described elsewhere (Fassati and Goff, 2001). The reaction mixture consisted of 100 mM Tris-HCl (pH 8.3; Sigma-Aldrich), 6 mM MgCl2 (Merck), 15 mM NaCl (Merck), 1 mM dithiothreitol (Merck), 2 mM dNTPs (Roche Diagnostics, Mannheim, Germany), 0.01% (v/v) Nonidet P-40 (Roche Diagnostics), and 50 μl of a purified viral sample (1 mg ml−1 protein), in a final volume of 100 μl. Three reactions were performed for each sample. The endogenous reactions were incubated at 37°C for 3 hr and then warmed to 75°C for 10 min. Viral DNA products were quantified by real-time PCR, using the FastStart DNA master SYBR green I kit (Roche Diagnostics) on a LightCycler instrument (Roche Diagnostics). The U5-R sequence of the HIV genome was amplified with the following primers: forward primer, 5′-GCT AAC TAG GGA ACC CAC-3′; reverse primer, 5′-GCT AGA GAT TTT CCA CAC TGA-3′. The R-U3 sequence of the genome was amplified with the following primers: forward primer, 5′-GAC CAA TGA CTT ACA AGG C-3′; reverse primer, 5′-AGC AGT GGG TTC CCT A-3′. When using the R-U3 primers the results will show only the decrease in viral capacity to perform first-strand transfer, because all the samples have been previously affected by the loss of capacity to perform the initiation step. Furthermore, the amount of DNA produced was linearly proportional to the quantity of virus added in the endogenous reaction, and the amount of viral RNA (template) present in the samples was not limiting.
Intracellular reverse transcription
Target 293T cells were seeded in 6-well plates (Nunc) at a density of 1 × 106 cells per well and incubated for 24 hr. Infections were carried out by replacing the medium with 500 μl of viral sample preincubated at 37°C for several periods of time; for each time point three wells were infected. Polybrene (8 μg ml−1) was added to the viral samples during the infection. The virus was allowed to adsorb to the cells for 4 hr, and then the cells were washed three times with PBS and fresh medium was added (Julias et al., 2001). Twenty hours after infection, the total intracellular viral DNA was isolated with a High Pure PCR template preparation kit (Roche Diagnostics) and eluted with 200 μl of elution buffer. The DNA products were quantified by the real-time PCR methods described previously, to quantify the U5-R and R-U3 DNA sequences.
Reverse transcriptase activity
To determine DNA polymerase activity a RetroSys RT activity kit (Innovagen, Lund, Sweden) was used. RT was released from the virions, using Triton X-100 (0.5% [v/v]; Sigma-Aldrich). The primer/template poly(rA)/poly(dT) was used and incorporation of bromodeoxyuridine followed. The product was quantified colorimetrically after incubation with the supplied phosphatase-conjugated antibody against bromodeoxyuridine. All reactions were linear with respect to incubation time and amount of virus added and three dilution sets were performed for each sample.
Viral genomic RNA degradation
To evaluate viral genomic RNA degradation an already established real-time RT-PCR method used for MLV vectors was modified (Carmo et al., 2004, 2008). Briefly, viral samples were incubated at 75°C for 10 min to release RNA and treated with DNase I (Sigma-Aldrich) to digest DNA derived from lysed cells. The viral RNA was transcribed into cDNA, using a first-strand cDNA synthesis kit (Roche Diagnostics) and a specific primer for the R-U3 sequence of the viral RNA (5′-AGC AGT GGG TTC CCT A-3′). The viral cDNA was quantified by real-time PCR using the following primers for the U5-R sequence of the viral genome: forward primer, 5′-GCT AAC TAG GGA ACC CAC-3′; reverse primer, 5′-GCT AGA GAT TTT CCA CAC TGA-3′.
To determine the amount of intact parental viral RNA inside the infected cells the protocol described previously to observe intracellular reverse transcription was used. Four hours after infection RNA was isolated from infected cells, using a High Pure viral nucleic acid kit (Roche Diagnostics), and eluted with 50 μl of elution buffer.
Entry of p24 protein
To determine the amount of p24 viral capsid protein inside the infected cells the protocol described previously to observe intracellular reverse transcription was used. In this method, cells were washed three times with PBS 4 hr after infection, trypsinized, and washed again with PBS. HIV p24 was liberated from infected cells by lysis with 1% (v/v) Nonidet P-40. The concentration of viral p24 protein was determined in an enzyme immunoassay (Innotest; Innogenetics, Ghent, Belgium) as recommended by the manufacturer.
Statistical analysis
Three complete sets of experiments were performed and a representative experiment is presented. For all the tests, data points presented are the average of at least three replicates of viral samples; the standard deviation is indicated in all figures. Half-lives (t 1/2) of the endogenous and intracellular reverse transcription processes, of RT activity, of viral RNA, and of infectivity were calculated by using linear regression analysis to fit the data in the linear range. Standard deviations were calculated from the regression analysis.
Results
Thermolability of reverse transcription process
The process of reverse transcription depends on specific interactions between the RT enzyme, the genomic RNA template/tRNA primer, as well as additional viral proteins. Synthesis of linear double-stranded proviral DNA requires several distinct steps: (1) initiation of (−)DNA synthesis, (2) first-strand transfer, (3) initiation of (+)DNA synthesis, (4) second-strand transfer, and (5) completion of a linear double-stranded DNA (Coffin et al., 1997; Abbink and Berkhout, 2008). We have shown that initial steps of the reverse transcription reaction, notably initiation of DNA synthesis and first-strand transfer, of MLV-derived gammaretroviral vectors are thermolabile and that this sensitivity determines the rate of retroviral infectivity decline (Carmo et al., 2008). We therefore tested the thermosensitivity of the reverse transcription reaction of lentiviral vectors. The thermosensitivity of two reverse transcription reactions, endogenous and intracellular, was compared (Fig. 1).
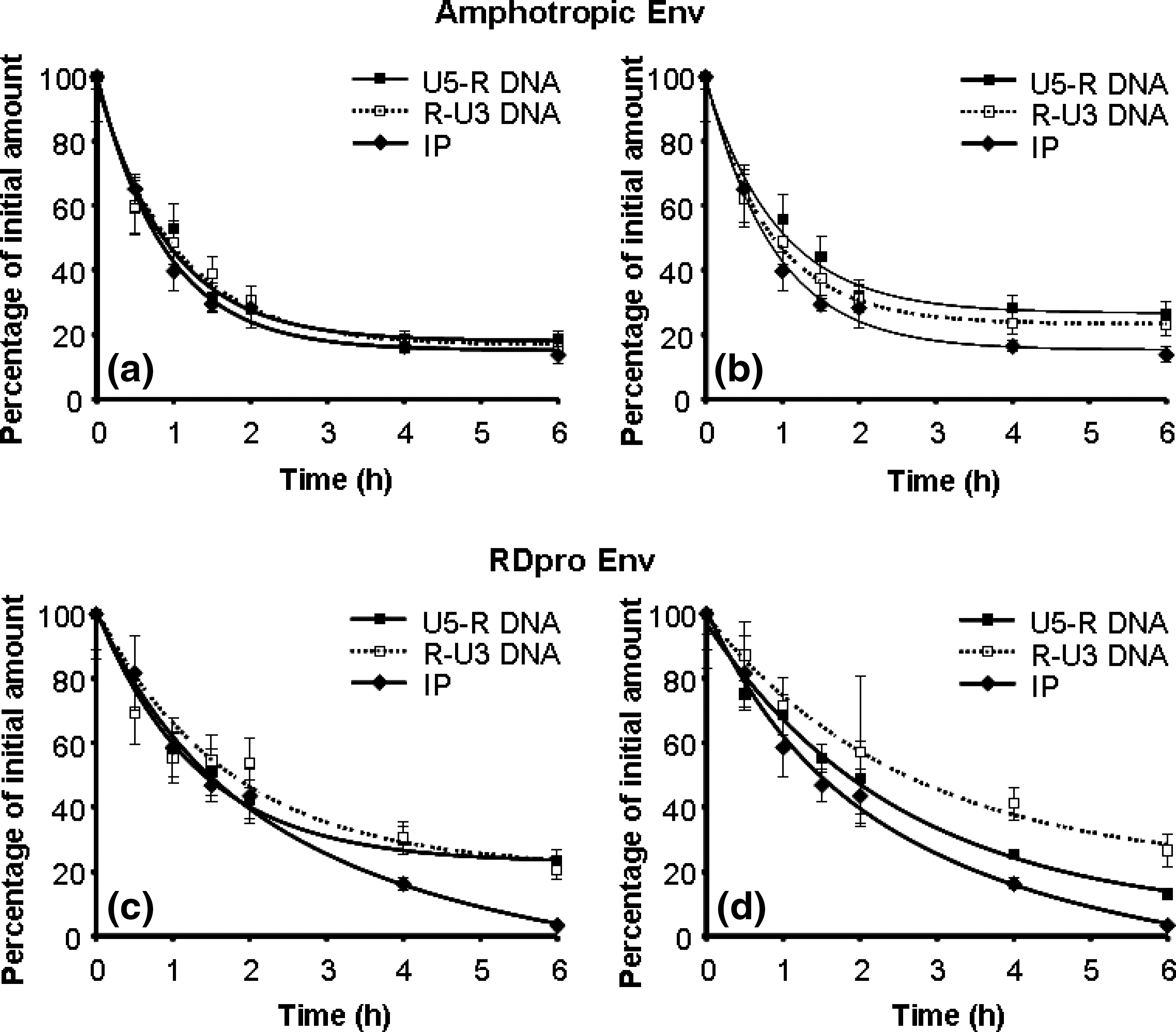
Correlation of viral DNA synthesis in the endogenous and intracellular reverse transcription reactions with viral infectivity, of lentiviral vectors with amphotropic Env (
In the endogenous reaction, purified vectors that were preincubated at 37°C were permeabilized with a nonionic detergent (Nonidet P-40) and incubated with deoxyribonucleotide triphosphate, thus enabling all steps of reverse transcription that culminate in the production of a functional proviral DNA, including the two long terminal repeats (LTRs) (Coffin et al., 1997). As purified viral samples, before the endogenous reaction, already contained small amounts of DNA, as reported previously (Trono, 1992; Chen et al., 2001), a control endogenous reaction without detergent was carried out. Under these conditions, dNTPs did not penetrate the viral envelope and the amount of DNA remained constant. The background DNA in virions, before the endogenous reaction, was subtracted from the amount of DNA obtained after the reaction.
In the intracellular reverse transcription reaction, viral samples that were preincubated at 37°C for various intervals were used to infect host cells and total DNA was extracted after 16 hr. By performing these two reactions in parallel it was possible to validate the data of reverse transcription thermosensitivity.
DNA products of the two initial steps of the reverse transcription process, that is, initiation of DNA synthesis, using primers for the U5-R sequence, and the first-strand transfer, using primers for the R-U3 sequence, were quantified in both reactions. For the lentiviral vectors with amphotropic envelope, the results obtained in the endogenous reactions are shown in Fig. 1a, and the results obtained for the intracellular reactions are depicted in Fig. 1b. It is clear from these two assays that the capacity of the virus to initiate the process of reverse transcription and to perform first-strand transfer declines rapidly by preincubation at 37°C. The half-life time (t 1/2) obtained from the endogenous reaction is 1.1 ± 0.1 hr for the initiation and 1.0 ± 0.2 hr for the first-strand transfer, and from the intracellular reaction t 1/2 was 1.3 ± 0.1 hr for the initiation and 1.0 ± 0.1 hr for the first-strand transfer. In a parallel experiment, viral infectivity was tested and the results indicated a rapid decline in infectivity after preincubation at 37°C, with a t 1/2 of 0.75 ± 0.03 hr, and a profile similar to that obtained for the two reverse transcription reactions (Fig 1a and b).
The tests described previously were also performed with lentiviral vectors pseudotyped with RDpro envelope. In Fig. 1c it is possible to observe the products of initiation and first-strand transfer of the endogenous reverse transcription and in Fig. 1d the products of initiation and first-strand transfer of the intracellular reverse transcription, together with the infectivity profile. The results confirm that the reverse transcription reaction is thermosensitive, with profiles of inactivation similar to that of infectivity loss. The t 1/2 values were 2.4 ± 0.5 and 2.7 ± 0.4 hr for the initiation and first-strand transfer of the endogenous reaction, respectively, and 2.0 ± 0.2 and 2.4 ± 0.2 hr for the initiation and first-strand transfer of the intracellular reaction, respectively. The decline in infectivity showed a t 1/2 of 1.3 ± 0.1 hr. Lentiviral vectors pseudotyped with ampho Env showed higher thermosensitivity in infectivity as compared with the RDpro vector. Interestingly, the higher thermosensitivity of the ampho Env vector was also observed in the reverse transcription process.
Thermolability of RT DNA polymerase activity and of viral RNA genome
For the synthesis of viral DNA several viral components need to be functional, including RT and the template viral RNA. Thus, the stability of these two viral components was analyzed (Fig. 2).

(
Loss of the DNA polymerase enzyme activity was observed after preincubation of the vectors at 37°C, using an exogenous template/primer [poly(rA)/oligo(dT)] (Fig. 2a). The decline rates of virion-associated DNA polymerase activity for lentiviral vectors pseudotyped with amphotropic envelope and with RDpro envelope were similar, with a t 1/2 higher than 10 hr for both vectors.
To evaluate the rate of viral RNA degradation in the virion, a method that specifically measures full-length viral genomic RNA, previously established for MLV-based gammaretroviral vectors, was adapted for lentiviral vectors (Carmo et al., 2008). Briefly, a primer with homology to the U3 region of lentivirus viral RNA was used for the RT step, such that the cDNA produced has only one R sequence, derived from the 5′ end of the viral RNA template. After cDNA synthesis a real-time PCR method was used to specifically amplify the cDNA R sequence at the 5′ end of the viral genome. Any internal nick within the genomic RNA template would result in the abortion of cDNA synthesis before the R sequence and thus produce a low signal in the real-time PCR.
The results, presented in Fig. 2b, show that the kinetics of genomic RNA degradation within virions preincubated at 37°C have a t 1/2 higher than 10 hr for both lentiviral pseudotypes.
Thermolability of RT DNA polymerase activity and of viral RNA genome of lentiviral vectors with VSV-G Env and without Env
To better understand the effect of Env on the stabilization of the viral particle it was necessary to verify the stability of viral components in vectors without Env. Thus, viral particles were produced by transient transfection without Env proteins. As a control for the transient transfection, vectors with VSV-G Env were also produced. The infectivity half-life of lentiviral vectors with VSV-G Env at 37°C was 1.3 ± 0.1 hr. Figure 3 shows the inactivation profiles of RT inactivation (Fig. 3a) and of viral RNA degradation (Fig. 3b) at 37°C of vectors with and without Env. Vectors without Env proteins present lower stability of both DNA polymerase activity and viral RNA. These results confirm the importance of the Env proteins in particle stabilization.
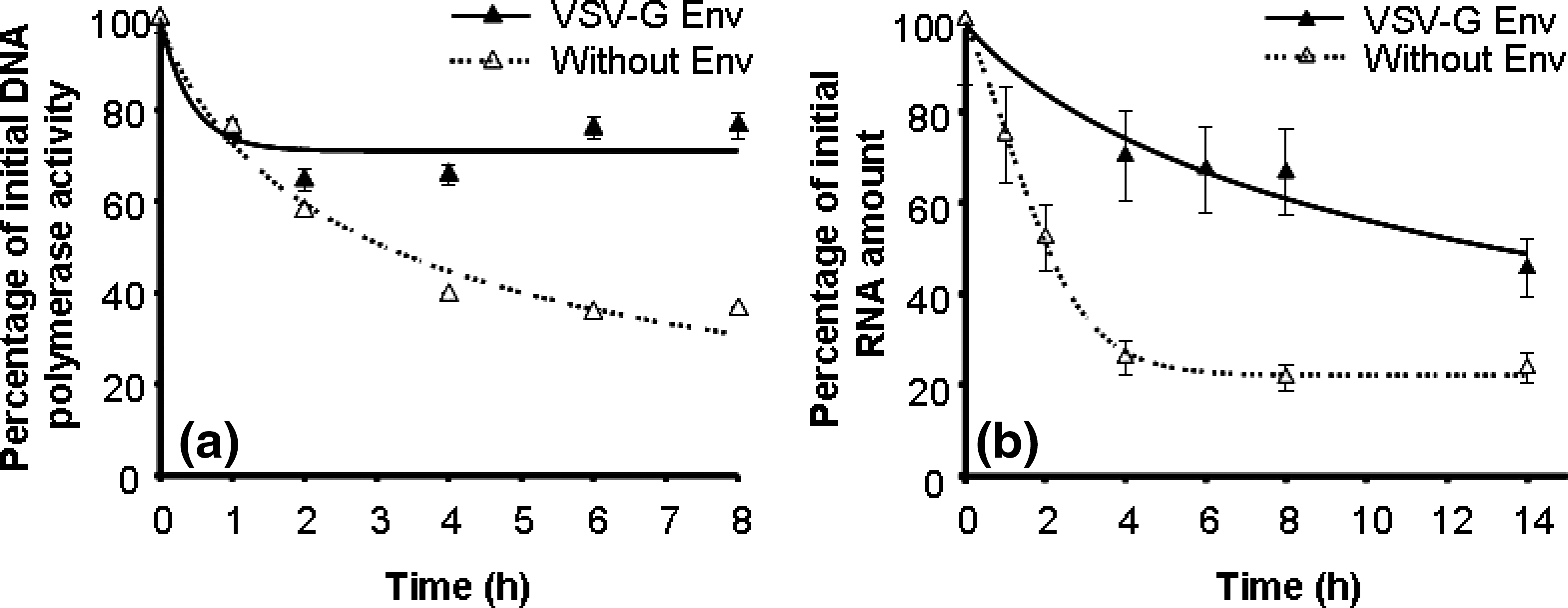
(
Thermosensitivity of viral entry to the host cell
As shown previously (Fig. 2b), viral RNA in the virion remains almost intact for more than 10 hr at 37°C, yet vectors preincubated at 37°C lost the capacity to perform synthesis of proviral DNA from the viral RNA. Thus, one needs to check whether the vectors lost their capacity to enter cells during incubation at 37°C. To this end, cells were infected with lentiviral vectors with either ampho Env or RDpro Env preincubated at 37°C; viral RNA was isolated from the cells 4 hr after infection and subjected to real-time RT-PCR analysis, to specifically measure levels of intact viral RNA in cells.
To validate this method, two experiments were performed. First, cells nonpermissive to lentiviral vectors with RDpro (NIH 3T3 cells; Rasko et al., 1999) were infected with these vectors in parallel to infection of 293T cells, and the protocol to quantify the viral RNA inside infected cells was followed. The results showed that only 10% of the vectors that entered 293T cells were detected in NIH 3T3 cells. These results demonstrate that the amount of viral RNA attached to cells (10%) does not affect the quantification of intracellular RNA in the permissive cells. Second, lentiviral vectors with or without envelope were used to infect 293T cells, and the protocol to quantify viral RNA inside infected cells was followed. The results have shown that less than 1% of the vectors without envelope entered the cells. We concluded, therefore, that the amount of viral RNA adsorbed to the cell surface after infection is low and can be controlled.
Intracellular viral RNA quantification after infection (Fig. 4a and b) indicated a slow decline due to the preincubation at 37°C. For both lentiviral vector pseudotypes, more than 50% of the intact viral RNA was still able to enter the host cells after 6 hr of preincubation.
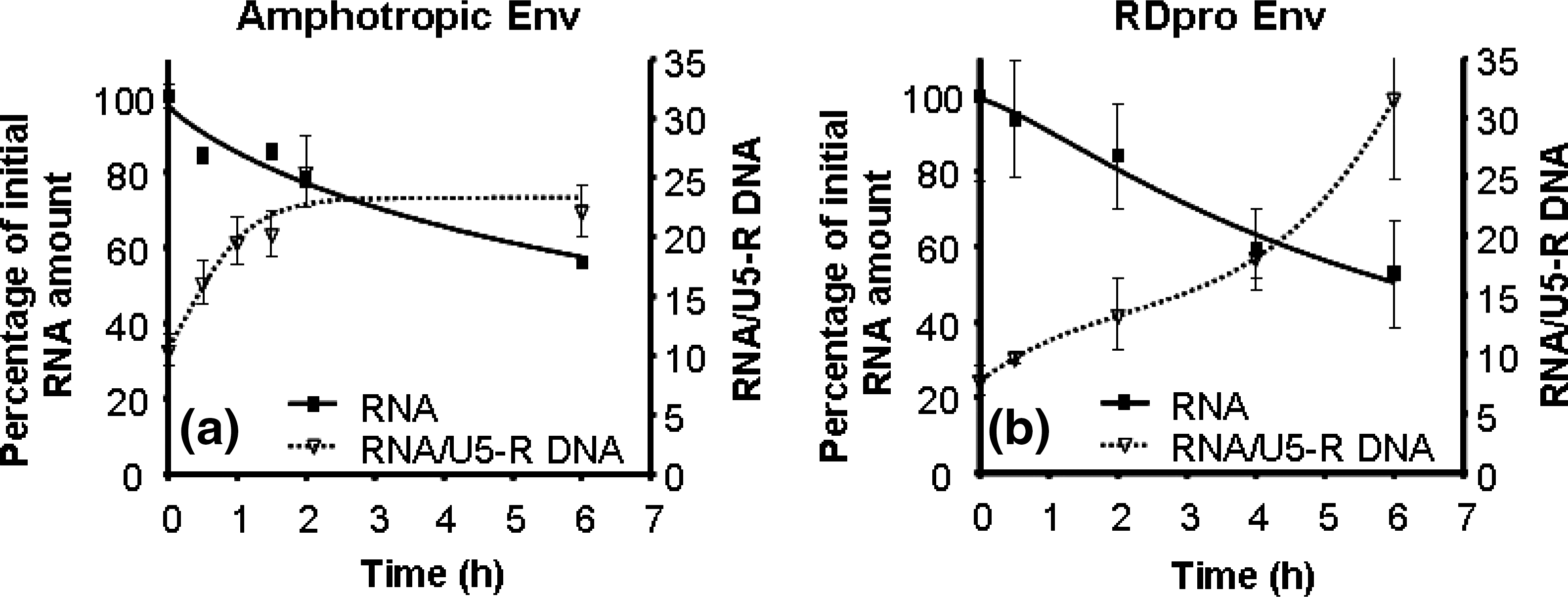
Viral RNA in the cytoplasm of cells infected with lentiviral vectors with amphotropic Env (
From these data, the ratio of intact viral RNA entering the cells versus the amount of DNA (U5-R) synthesized within the cells was calculated for each preincubated viral sample; this ratio indicates the efficiency of the reverse transcriptase machinery to synthesize DNA from viral RNA. About 10 copies of viral RNA give rise to 1 copy of viral DNA. As shown in Fig. 4a and b, the ratio of template RNA to product DNA, after infection, increased with time for both lentiviral vector pseudotypes. This shows that an increase in RNA that is not reverse transcribed into DNA occurred during the time of incubation. Nevertheless, for lentiviral vectors with ampho Env after 2 hr of preincubation, a 2-fold increase was observed in the RNA/U5-R DNA ratio, and for vectors with RDpro Env, only after 4 hr was a 2-fold increase verified.
To validate that the amount of intact viral RNA in the host cell could be used as a measure of viral entry, the amount of p24 viral capsid protein in the preincubated viral samples that enters the host cells was quantified. Cells were infected with viral samples preincubated at 37°C for several periods of time (the same samples used for verifying the amount of intact RNA entering the cells); 4 hr after infection cells were extensively washed, trypsinized, and lysed and the amount of p24 protein was quantified (Fig. 5).
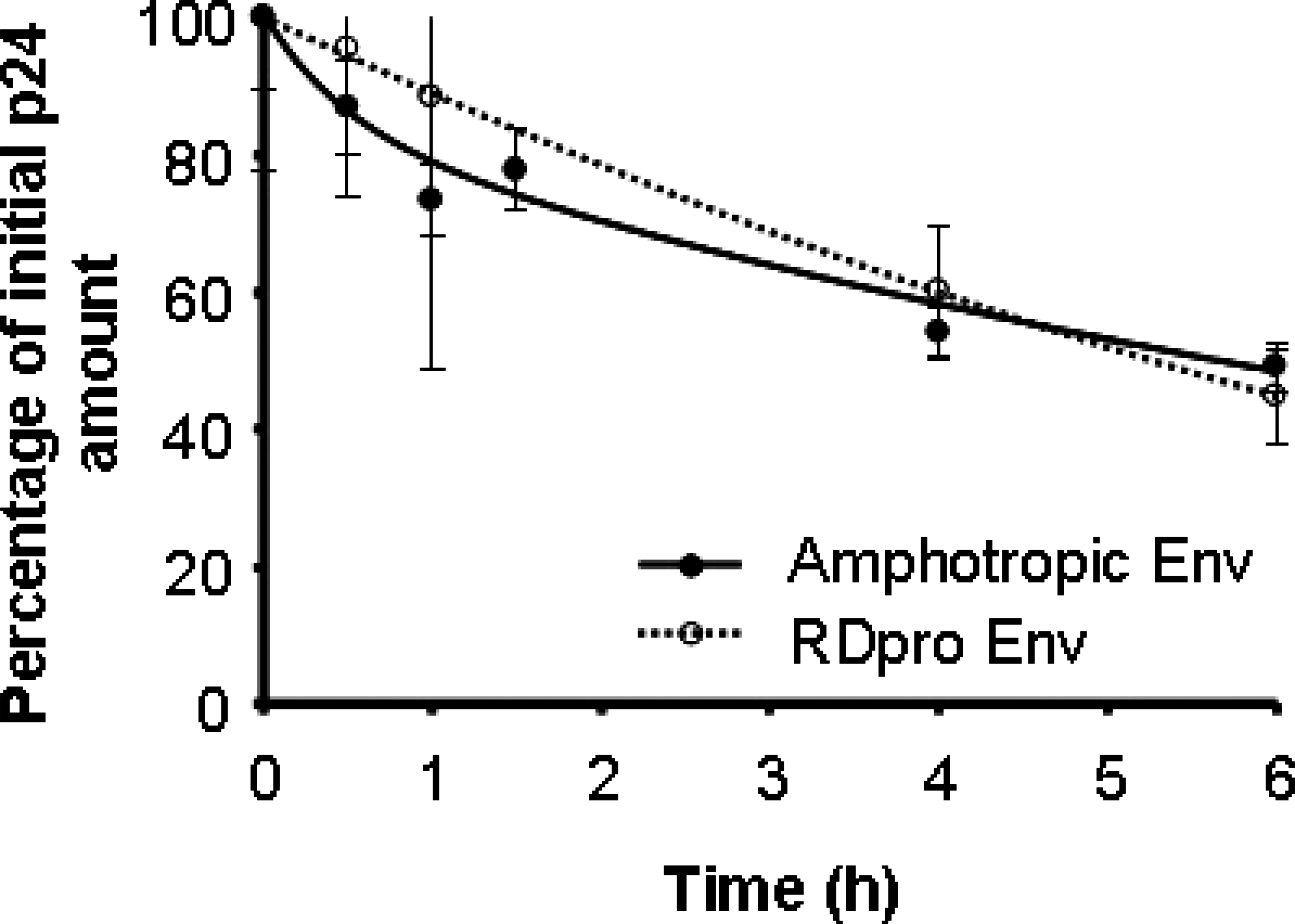
Viral p24 proteins in the cytoplasm of cells infected with lentiviral vectors with amphotropic Env and with lentiviral vectors with RDpro Env, preincubated at 37°C. The initial p24 concentrations, in cells infected with viral samples before incubation at 37°C, were taken as 100%.
The results show that the amount of p24 protein that entered cells declined slowly due to preincubation at 37°C; at 6 hr of preincubation, 50% of the p24 proteins still entered the cells, for both lentiviral vectors, confirming the results of viral RNA entry.
Discussion
Lentiviral vectors are a powerful tool for gene transfer and gene therapy applications. However, several hurdles exist in the large-scale production and efficient application of lentiviral vectors. The low thermostability of the virus affects the final yield of vectors produced, as well as the quality/efficacy of the preparations to be used in clinical trials. Hence, a better knowledge of how lentiviral vectors lose infectivity is necessary in order to devise strategies for vector stabilization. Our laboratory showed that the main mechanism of MLV-based gammaretroviral vector infectivity loss was related to the thermosensitivity of the reverse transcription process (Carmo et al., 2008). Because of the similarity of gammaretrovirus and lentivirus, the sensitivity of the reverse transcription process at 37°C was studied as a possible mechanism of lentiviral vector inactivation; vectors pseudotyped with two different Env proteins (amphotropic and RDpro) were used.
The results indicated that for both lentiviral vector pseudotypes the capacity to perform reverse transcription at 37°C decreases rapidly and with high correlation to the vector infectivity loss (Fig. 1). Similar thermosensitivity was obtained for the endogenous and intracellular reverse transcription reactions. Two distinct steps of the reverse transcription process were found to be thermolabile: initiation of (−)DNA synthesis and first-strand transfer. The observations with lentiviral vectors are comparable to those obtained for gammaretroviral vectors, in terms of the inactivation mechanism (Carmo et al., 2008). It was also observed that neither RT enzyme inactivation nor viral RNA degradation has a role in the loss of viral capacity to perform reverse transcription, because these two viral components are rather stable at 37°C (Fig. 2). It should be noted that RT and RNA from lentiviral vectors showed higher stability as compared with gammaretroviral vectors. It is known that RTs from gammaretroviral and lentiviral vectors have different structures—lentiviral RT is a dimer of two different subunits and both contain DNA polymerase activity, whereas gammaretroviral RT is a monomer (Coffin et al., 1997)—which may play a role in their different thermostabilities. The effect of preincubation at 37°C on viral entry into the host cell was studied by measuring the amount of intact viral RNA and of p24 capsid protein that entered the cells (Figs. 4 and 5). As the amount of these two viral components after cell entry declined at a slower rate, when compared with viral infectivity, it follows that entry of viral RNA into the cell is not a rate-limiting factor for infectivity decline. Because the ratio of intact viral RNA to the amount of viral DNA produced after infection increased during the preincubation time, it is possible to infer that vectors lose their capacity to transcribe RNA into DNA and that the amount of RNA inside the cell does not restrict reverse transcription. Thus, the first conclusion drawn from this work is that the loss of viral capacity to perform reverse transcription is a main mechanism of lentiviral vector inactivation. With RT inactivation, RNA degradation and loss of capacity of the virus to enter the host cells have only a small role in vector inactivation.
An overview of the tests performed in the present study is presented in Fig. 6, in addition to the thermosensitivity (t 1/2) of each step. It is clear that preincubation of virus at 37°C affects the infection process after the entry step for both lentiviral pseudotypes. Vectors pseudotyped with RDpro Env are more stable than ampho Env pseudotyped vectors with respect to viral infectivity and, interestingly, this higher stability is also observed in the reverse transcription process. In vectors with RDpro Env the limiting thermolabile step is the initiation of DNA synthesis and not the first-strand transfer, as observed for vectors pseudotyped with amphotropic Env and as previously shown for gammaretroviral MLV vectors (Carmo et al., 2008).
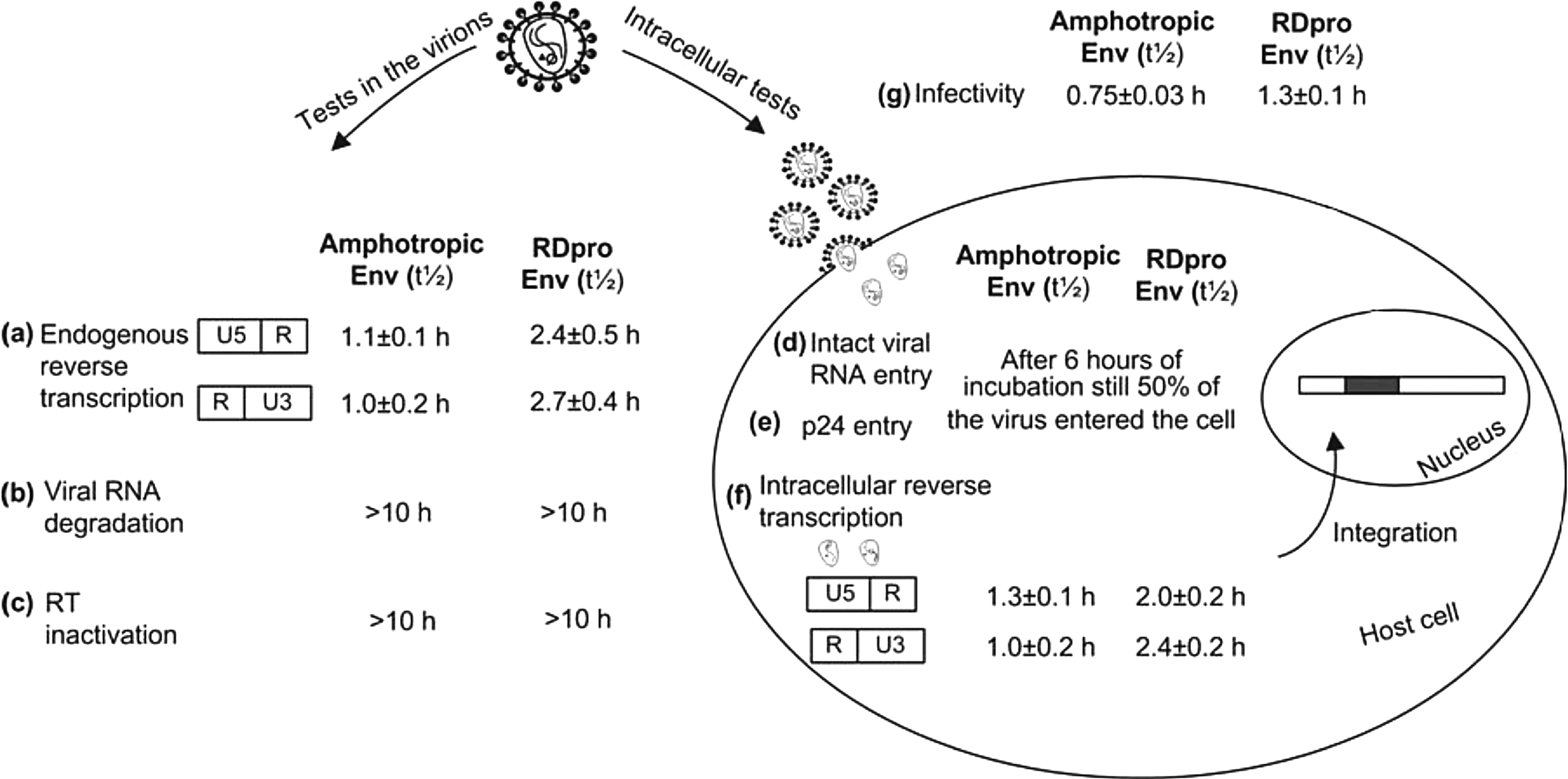
Overview of the tests performed in the analysis of lentiviral thermosensitivity and half-lives obtained in each test for vectors with amphotropic Env and for vectors with RDpro Env. Viral samples were preincubated at 37°C for several periods of time and tested for (
It is worth noting that the higher stability of vectors with RDpro Env was not reflected by different rates of RT enzyme inactivation or by the rate of RNA degradation. Also, the amount of viral RNA and of viral p24 protein that entered the host cells declined at similar rates for both pseudotype vectors. Nevertheless, the ratio of intact viral RNA to the amount of viral DNA produced after infection increased at different rates for the two pseudotyped vectors, with Ampho Env showing a faster increase. Thus, the second conclusion that is drawn from this work is that lentiviral vector pseudotyped with RDpro Env is more stable than vector with ampho Env and that higher viral stability is due to a slower loss of the capacity to perform reverse transcription. To verify the stabilization of Env proteins on the viral particle constituents, vectors without Env proteins were produced. The results have shown that these vectors present lower stability of both RT and viral RNA, confirming that the Env proteins confer protection to the viral constituents (Fig. 3). The stability of RT and viral RNA was also studied in lentiviral vectors with VSV-G Env as a control for transient transfection. These vectors present the same stability of RT and viral RNA as lentiviral vectors with Ampho Env and with RDpro Env, as well as in terms of infectivity. Unfortunately, because vectors with VSV-G Env and vectors without Env were produced transiently it was not possible to obtain consistent results from the endogenous reverse transcription between different productions. The vector samples produced by transient transfection are highly contaminated with plasmid DNA and production by transient transfection presented high variability.
It was also possible to observe in this work that the efficiency in starting reverse transcription was lower for lentiviral vectors than for gammaretroviral vectors. This efficiency is indicated by the RNA/U5-DNA ratio at zero time of preincubation. In lentiviral vectors 1 copy of U5-DNA is synthesized per 10 copies of viral RNA that enter the cell whereas in gammaretroviral vectors the value is 1 per 5 copies (Carmo et al., 2008).
The reverse transcription process occurs in the cytoplasm in a complex that includes viral RNA, tRNA, reverse transcriptase, nucleocapsid proteins, and integrase. The integrity of this complex is of the utmost importance for the completion of proviral DNA synthesis (Gotte et al., 1999; Fassati and Goff, 2001; Nermut and Fassati, 2003). In this work it was observed that neither viral RNA nor RT suffers inactivation at 37°C, and thus other components should be thermosensitive. The nucleocapsid protein (NC) plays an essential role in several steps of the reverse transcription process. It unwinds viral RNA, facilitates the strand transfers, and improves the processivity of the DNA polymerase (Gotte et al., 1999; Nisole and Saib, 2004; Thomas and Gorelick, 2008). It is possible that at 37°C the native NC loses the conformation required for a functional reverse transcription complex, leading to inactivation of reverse transcription. Another important constituent of the reverse transcription complex is the primer tRNA; its proper interaction with the reverse transcriptase enzyme and the template RNA influences the initiation of the reverse transcription process (Gotte et al., 1999). Further studies on the stability of these two components at 37°C, as well as the stability of their interactions with RT and template RNA, should be pursued to assess the cause of the fast inactivation of the reverse transcription complex.
Two different envelopes were used in this work for pseudotyping lentivirus, amphotropic and RDpro; both derive from gammaretroviruses. The ampho Env (derived from murine leukemia virus) efficiently pseudotypes lentiviral vectors whereas the RD114 Env (derived from feline endogenous virus) has a low capacity to pseudotype lentiviral vectors (Sandrin et al., 2002). Consequently, RD114 Env was engineered to be used in lentiviral vectors, by substitution of the cytoplasmic tail with that of MLV Env (Sandrin et al., 2004). Furthermore, the R peptide cleavage site sequence was replaced with that of a matrix–capsid cleavage site in HIV Gag to create RDpro (Strang et al., 2004). It was shown that Env protects viral constituents from inactivation/degradation at 37°C. Thus, a small change in Env can lead to a change in overall vector stability, by providing higher protection to the reverse transcription complex.
The findings reported in the present study have profound practical relevance, as stabilization of the reverse transcription process should be pursued to improve the stability of lentiviral vectors, with one possible solution for stabilization being modification of the Env protein. This has important implications for the production and storage of lentiviral vectors and consequently for their use as gene therapy vectors.
Footnotes
Acknowledgments
The authors acknowledge financial support received from the Fundação para a Ciência e Tecnologia-Portugal (THERAVECT, FCT/POCI/v.5/A003/2005 and POCTI SFRH/BD/18239/2004) and the European Commission (Clinigene Network of Excellence, LSHB-CT-2006-018933).
Author Disclosure Statement
No competing financial interest exists.