
Abstract
Detection of nonselective adenoviruses in tissue- or tumor-selective oncolytic adenovirus preparations presents a technical challenge because of the conditionally replication-competent nature of oncolytic adenoviruses. Although quantitative PCR has been used extensively for detecting specific genes that are likely present in nonselective recombinants, the actual biological activity of nonselective genetic recombinants has not been demonstrated. Therefore, a bioassay that amplifies nonselective adenoviruses through multiple passages in nonpermissive cells was developed to detect biologically active nonselective recombinants using CG7870, a prostate-specific oncolytic adenovirus. The assay was sensitive, and its results were consistent with a quantitative PCR assay for four lots of CG7870. CG0070, a pan-tumor oncolytic adenovirus with no detectable wild-type–like recombinants by PCR, was subjected to a variation of this bioamplification assay using two different nonpermissive cell lines to both verify PCR results and assess its genetic stability under selection pressure. No evidence of the presence of biologically active nonselective recombinants was seen in the original material or after serial passaging in nonpermissive cells. Thus, this bioamplification assay is able to detect nonselective recombinants, and its results are consistent with quantitative PCR assays. A modified version of this assay is also useful for assessing the genetic stability of oncolytic adenoviruses that have no PCR-detectable recombinants.
Introduction
Due to the conditionally replication-competent nature of the oncolytic adenovirus, and as all tissue- or tumor-specific promoters appear to have a basal level of activity even in nonpermissive cells, albeit at a very low level, it may be difficult to detect the biological activity of replication-competent recombinants that have a low incidence of occurrence against the background noise of the comparatively massive amounts of oncolytic adenovirus in a preparation. One characteristic of oncolytic adenoviruses is that they preferentially replicate in tumor cells compared with normal cells (Dalba et al., 2005; Ko et al., 2005). The undesired wild-type–like nonselective recombinant adenovirus will replicate equally well in both tumor and normal cells, which suggests that leveraging the differential replication of recombinants in normal cells may provide a rational strategy for developing a bioassay to distinguish wild-type–like recombinants. WI-38 cells, normal diploid lung fibroblasts derived from embryonic lung tissue (Hayflick and Moorhead, 1961), were chosen for use as the normal cell control after extensive screening.
CG7870 is an oncolytic adenovirus generated by replacing the E1A promoter upstream of the E1A region with the rat probasin promoter, and replacing the E1B promoter upstream of the E1B region with the prostate-specific antigen (PSA) promoter and enhancer elements (Dilley et al., 2005; Small et al., 2006) (Table 1). These two inserted promoters make CG7870 highly selective for prostate tissue. Like many adenovirus vectors, CG7870 was produced in human embryonic kidney 293 (HEK293) cells, because this cell line provides the E1 region of human adenovirus type 5 (Ad5) genome (Graham et al., 1977) necessary for the production process. However, the presence of Ad5 E1 sequence in HEK293 cells also provides the molecular basis for the potential generation of recombinants that have regained the wild-type E1 promoter sequences during replication in HEK293 cells (Lochmuller et al., 1994; Hehir et al., 1996). A quantitative PCR (qPCR) assay that is specific for the E1 promoter sequence revealed that most CG7870 production lots had a low level of wild-type genomes, ranging from one wild-type genome in 1 × 107 to one in 2 × 108 genomic copies of CG7870. Whether the wild-type genomic copies detected in the lots were biologically active recombinant adenoviruses that could replicate in normal cells had not been established. Thus, a bioamplification assay that serially passaged oncolytic adenovirus on WI-38 cells was developed for CG7870 lots to determine if the recombinants with wild-type E1 promoters were biologically active and potentially nonselective for prostate cells.
The oncolytic adenovirus CG0070 was derived from Ad5 with the following modifications (Ramesh et al., 2006): (a) the E1A promoter was replaced with the E2F-1 promoter, (b) the E3 gp19kD–coding region was replaced with human cDNA for granulocyte-macrophage colony stimulating factor (GM-CSF), and (c) a polyadenylation signal was inserted upstream of the E2F-1 promoter to prevent unregulated expression of E1A (Table1). With the E2F-1 promoter, CG0070 is targeted to preferentially replicate in tumor cells with mutations of retinoblastoma protein (Rb), eventually leading to tumor cell lysis and dispersal of progeny virions to infect and replicate in surrounding tumor cells. The efficacy of CG0070 is expected to be further enhanced by the secretion of GM-CSF, which may stimulate a tumor-specific immune response against distant metastases (Ramesh et al., 2006). A qPCR method specific for the wild-type E1A promoter, with a sensitivity of five copies of wild-type E1A sequence in a background of 5 × 109 copies of CG0070 genome (unpublished data), was used to test all CG0070 lots. No wild-type recombinants were found in any lot of CG0070. These results were expected, because CG0070 was produced in HeLa-S3 cells, which produce the Rb protein necessary for virus production, but are free of wild-type adenovirus sequences and do not, therefore, support recombination to a wild-type genome. In this case, the concern was whether CG0070 would go through genetic recombination during manufacturing processes and generate nonselective recombinants that do not contain the wild-type E1A promoter and, therefore, are not detectable by the qPCR assay specific to Ad5 E1A promoter. For a complete assessment of the genetic stability of CG0070, the adenovirus was tested with the bioamplification assay developed for CG7870 but using two cell lines, WI-38 and MRC-5, to increase the probability of detecting any nonselective recombinants.
Here we describe a bioamplification assay for detecting wild-type–like and nonselective recombinants for oncolytic adenoviruses, in which oncolytic adenoviral vectors are serially passaged on nonpermissive cells to preferentially amplify any existing and biologically active nonselective recombinants and to favor generation of new nonselective recombinants. The resulting amplified progeny virus populations were quantified and analyzed for biological and genetic properties of any emerging recombinants using a battery of analytical methods. The results of this assay for two oncolytic adenoviruses, CG7870 and CG0070, are presented as case studies for applying the bioamplification assay in the characterization of oncolytic adenovirus preparations.
Materials and Methods
Viruses
CG7870 was produced in HEK293 cells (Microbix, Toronto, ON, Canada), purified by anion-exchange chromatography, and formulated in 5% sucrose, 0.05% Tween-80, 10 mM Tris, 1 mM MgCl2, 1% glycine, pH 7.8, by Cell Genesys as described (Yu et al., 1999). CG0070 was produced in HeLa-S3 cells (Ramesh et al., 2006), purified, and formulated as described for CG7870. Wild-type human Ad5 (Yu et al., 1999), and CG8840, an oncolytic adenovirus selective for bladder urothelium cells (Zhang et al., 2002), were produced and formulated as described for CG7870. All virus preparations were stored below –60°C until use.
Cells
WI-38 cells (ATCC, Manassas, VA; catalog no. CCL-75) were cultured in RPMI 1640 medium with 2 mM L-glutamine, 1% penicillin–streptomycin, and 10% fetal bovine serum (FBS; Hyclone, Thermo Fisher Scientific, Pittsburgh, PA). MRC-5 cells (ATCC, catalog no. CCL-171) were cultured in Dulbecco's minimum essential medium high glucose with 2 mM L-glutamine, 1.0% penicillin–streptomycin, and 10% FBS. Both cell lines were subcultured twice a week. LNCaP cells (ATCC, catalog no. CRL-1740) were cultured in RPMI 1640 containing 10% FBS and 4 mM GlutaMax (Invitrogen, Carlsbad, CA). HEK293 cells were cultured in serum-free CD-293 medium (Invitrogen) supplemented with 4 mM L-glutamine (Invitrogen). HepG2, LOVO, MCF-7, OVCAR-3, ARPE-19, and IMR-90 cell lines were all from ATCC and cultured as for WI-38 cells.
Virus yield assay
The virus yield in LNCaP, PC-3, WI-38, HepG2, LOVO, MCF-7, OVCAR-3, ARPE-19, IMR-90, and lung fibroblast cells was determined as described by Dilley et al. (2005) for CG7870, CG8840, and Ad5. In brief, each cell line was seeded at 4 × 104 cells per well in a six-well plate and infected with the virus at the specified multiplicity of infection (MOI) 24 hr later. Three days after infection, the plates were subjected to three freeze-and-thaw cycles to release progeny virus from cells. Each supernatant was tested by a standard plaque assay using HEK293 cells for the virus yield expressed as plaque-forming units per milliliter (pfu/ml).
CG7870 bioamplification assay
WI-38 cells were seeded into T-175 culture flasks at 1 × 106 cells per flask in 30 ml of culture medium and passed to new flasks when reaching 90% confluence in approximately 1 week. One week prior to initiation of the assay, WI-38 cells were harvested and 3.5 × 107 total viable cells were seeded into one 10-chambered Corning Cell Stack (Thermo Fisher Scientific) containing 1.5 L of culture medium in order to supply sufficient cells for each assay. Culture of WI-38 cells was limited to eight total passages from thawing to assay use due to the concern of cell senescence. On the first day of the assay, WI-38 cells were harvested from the Cell Stack and seeded into 20 T-300 flasks at 6 × 106 total viable cells per flask in 20 ml of infection medium (RPMI 1640 containing 2% FBS, 4 mM GlutaMax, and 1% penicillin–streptomycin). Cells were infected with 3 × 109 adenovirus particles per T-300 flask, giving an MOI of 500 virus particles (vp)/cell. Flasks were incubated undisturbed at 37°C, 5% CO2 for 24 hr, then gently rocked inside incubators for an additional 24 hr. Following 48-hr total incubation, the inoculum was replaced with 30 ml of fresh culture medium containing 10% FBS. Five days after refeeding, flasks were treated with three freeze-and-thaw cycles between −80°C and 37°C to release virus. Cells adhering to the flask surface were dislodged by scraping and combined with the culture supernatant. The lysate was clarified at 1,000 × g centrifugation for 10 min and stored at −80°C. For the second passage, a freshly seeded T-300 flask of WI-38 cells containing 10 ml of infection medium was inoculated with 10 ml of clarified supernatant, one third of total lysate from previous passage. Cultures were incubated and harvested as described for the primary amplification passage. The assay was normally conducted with four total passage cycles with 20 flasks in each cycle. Cell lysate from each harvested flask was analyzed without pooling. The cell lysate from each flask at each passage was assayed for yield of CG7870 and analyzed for wild-type virus or wild-type–like recombinants by qPCR. The first passage was infected with a fixed MOI (500 vp/cell), but subsequent passages were infected with one third of the cell lysate in order to maintain a similar MOI range throughout the assay based on the PCR quantification of CG7870 genomic copies. Amplified materials from selected passages were tested for their ability to replicate and kill WI-38 and LNCaP cells. Cell lysate from each flask of the fourth passage of each sample was further analyzed by restriction enzyme mapping of genomic DNA extracted from purified adenovirus particles to identify any distinguishable virus subpopulation(s).
CG0070 bioamplification assay
WI-38 and MRC-5 cells were cultured in T-175 flasks for the first week after thawing and expanded to T-300 flasks using the media and culture conditions described above. CG0070 was serially passaged on WI-38 cells and MRC-5 cells as described for CG7870 with the following exceptions. The first infection cycle was started with five flasks, and 10 ml of the cell lysate harvested from each flask was then passaged to each of two new flasks in the subsequent cycle. At the end of the four passages, a total of 40 flasks were available for analysis for each cell line.
CG7870-specific qPCR assay
Adenoviral DNA was isolated from 200 μl of the clarified cell lysate from each flask using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA) per the manufacturer's instructions. DNA was eluted from purification columns in a 55-μl volume, 5 μl of which was analyzed by a TaqMan (Applied Biosystems, Foster City, CA) qPCR assay specific for CG7870. This assay produced a 100-base pair (bp) amplicon from a forward primer complementary to the E1A promoter region (CG7870 nucleotides [nt] 520–539) and a reverse primer to the probasin promoter (CG7870 nt 601–620). The 26-bp probe hybridizes to the junction of the E1A and rat probasin promoter (CG7870 nt 541–566). PCR reactions (50 μl) were composed of 1 × Universal Master Mix, 100 nM dNTPs, 400 nM forward and reverse primers (IDT, Coralville, IA), and 100 nM probe (Applied Biosystems, Carlsbad, CA). Amplifications were conducted using PCR cycling conditions of 50°C for 2 min, 95°C for 10 min, and 40 cycles alternating between 95°C for 15 sec and 60°C for 1 min.
Wild-type Ad5 long amplicon quantitative PCR assay (LA-qPCR)
CG7870 DNA was extracted and purified as described above and analyzed using a qPCR primer/probe combination specific for wild-type adenovirus. This assay was designed to produce a 3,964-bp amplicon using a forward primer specific for the junction between the E1A promoter and gene (Ad5 nt 538–555), a probe complementary to sequences in E1A (Ad5 nt 574–594), and a reverse primer that hybridizes in E2B (Ad5 nt 4379–4402). This assay is specific for wild-type adenovirus because CG7870 fails to hybridize with the forward primer due to its E1A modifications. Because the reverse primer does not overlap with the Ad5 sequence (Ad5 nt 1–4344) incorporated into HEK293 host cell DNA (Hehir et al., 1996), residual host cell DNA would not be amplified and quantified in this assay. Five microliters of DNA extracted from cell lysates was incorporated into each PCR reaction (50 μl total) composed of 1 × PCR Buffer, 100 nM dNTPs, 400 nM forward and reverse primers, 100 nM probe, 2.5 units of L(ong)A(mplicon) Taq polymerase (TaKaRa Bio, Madison, WI), and 1 × ROX dye (Invitrogen). Amplifications were conducted using PCR cycling conditions of 94°C for 1 min, and 40 cycles alternating between 94°C for 15 sec and 68°C for 4 min. This long extension time was necessary for adequate amplification of such a large amplicon.
Poisson distribution
One unit of the detectable analyte is defined as the detectable biological activity of the wild-type viruses or wild-type–like recombinants. The number of units per flask was calculated assuming that the number of positive events in the bioamplification assay follows a Poisson distribution P(x; λ) = (e -λ)(λ x )/x! where e is the base of the natural logarithm (2.718); x is the number of occurrences of an event; x! is the factorial of x; and λ is the expected number of occurrences. The number of negative flasks (p 0) can then be used to calculate the average units per flask (α) by the equation α = ln (p o /N), where N is the total number of flasks.
Potency assay for amplified CG7870
Potency assays were performed on both LNCaP and WI-38 cells for the amplified CG7870 materials. Each cell line was seeded at 1 × 104 total viable cells per well in 96-well plates in RPMI 1640 containing 10% FBS, 4 mM L-glutamine (and 1% penicillin–streptomycin for WI-38 cells only). CG7870 materials were serially diluted in RPMI 1640 containing 4 mM L-glutamine, and titrated based on particle titers (CG7870 starting material) or CG7870 genome copies determined by the CG7870-specific qPCR (passage material) to make final MOIs from 100,000 to 0.024 vp or genome copies per cell in each well (four replicate wells for purified virus preparations, two replicates for passage material). Plates were incubated at 37°C, 5% CO2 for 7 days before addition and incubation of alamarBlue (Invitrogen) for 8 hr. Plates were read with a fluorescent plate reader (Molecular Devices, Sunnyvale, CA) using a 544-nm excitation wavelength light source and a 590-nm emission wavelength detector. Fluorescence data were analyzed using SoftMax Pro (v. 3.1.1). Ad5 was used as a positive control for nonselective adenovirus at the same MOI.
Potency assay for amplified CG0070
The potencies of CG0070 and amplified materials from each passage were evaluated in both WI-38 and MRC-5 cells. The infection and incubation conditions were identical to those for CG7870 except for the final assay readout. Cell viability on day 7 postinfection was evaluated by the addition of MTS solution (Promega, Madison, WI), and the formazan product was measured after 1-hr incubation at 37°C in 5% CO2 at 490 nm with a plate reader (Molecular Devices).
Restriction enzyme digest of amplified CG7870 DNA
To generate sufficient adenovirus particles for the restriction digest, clarified cell lysate (1 ml total) from each of the fourth passage flasks was used to infect 50 ml of suspension-adapted HEK293 cells (5 × 105 cells/ml) at MOI 50 genome copies per cell. Each was harvested 48–72 hr postinfection, and cell lysates were prepared as described for bioamplification. Clarified lysates were applied to Vivapure Q columns (VWR, West Chester, PA) to isolate viral particles following the manufacturer's instructions. A QIAamp DNA Blood Mini Kit (Qiagen) was used to extract adenoviral DNA. Isolated DNA was repurified by ethanol precipitation, resuspended in water to a final concentration of 50 ng/μl, and stored at –20°C prior to analysis. In each digestion reaction (10 μl total), 250 to 500 ng total DNA was treated with BspHI and BsrGI combined (5 units each), DraI (10 units), or ScaI (20 units) following the manufacturer's instructions (New England Biolabs, Ipswich, MA). The reaction mixture was diluted with gel-loading buffer (Blue Juice; Invitrogen) and loaded onto a 0.8% SeaKem Gold agarose gel (25 cm total length; Lonza, Rockland, ME) in TBE (Tris borate EDTA; Invitrogen). Molecular weight markers, Marker II and Marker III (Roche Diagnostics, Indianapolis, IN), were included on the same gels. Electrophoresis was conducted at 2 to 2.5 volts/cm, the DNA bands were stained with SybrGold nucleic acid stain (Invitrogen) in 1 × TBE for 40 min, and digitally documented using an Alpha Innotech Fluorchem 8900 imaging system (Cell Biosciences, Santa Clara, CA) with an FLSC-500 filter (excitation at 302 nm).
PCR assays for amplified CG0070 DNA
The adenoviral DNA extracted for the restriction enzyme digestion of CG0070 was also tested in three PCR assays. The first PCR reaction, which amplified the entire E2F-1 promoter, used a forward primer specific to Ad5 nt 232–260 and a reverse primer for Ad5 nt 881–912 (same nucleotides as CG0070 nt 1179–1210), which generates an amplicon of 979 bp for CG0070. The second PCR had a forward primer specific to Ad5 nt 28511–28529 (identical to CG0070 nt 28809–28827) and a reverse primer (5’-GTCTGTAGGCAGGTCGGCTC-3’) that hybridized to the 5’ end of inserted human GM-CSF gene located in CG0070 nt 29240–29259. The third PCR reaction used a forward primer specific to the 3’ end of the inserted human GM-CSF gene (5’-GCCTCACCAAGCTCAAGGG-3’) located in CG0070 nt 22295–29313 and a reverse primer against the Ad5 nt 29370–29389 (same as CG0070 nt 29626–29645). The second and third PCR reactions would generate amplicons of 451 bp and 351 bp, respectively, for CG0070. PCR reactions were performed in a total volume of 25 μl using a HotstarTaq Master Mix Kit (Qiagen), 10 pmol of each primer, 50 ng of DNA, and 5 μl of Q-Solution (Qiagen). Standard PCR reactions were performed in a GeneAMP PCR System 9700 (Applied Biosystems) with 40 cycles of 0.5 min at 95°C, 0.5 min at 60°C, and 1.5 min at 72°C after activation for 10 min at 95°C. The first PCR products were analyzed by gel electrophoresis in 1.0% agarose in TBE and premixed with ethidium bromide; the second and third PCR products were analyzed on a 2% agarose gel. The 100-bp DNA ladder (Invitrogen) was included in each gel.
Results
Screening for a suitable cell line for bioamplification
Production test samples would likely be comprised of relatively few wild-type or nonselective recombinants within a high background of the oncolytic adenovirus product. Therefore, the first challenge in developing this assay was to identify a cell line that was least permissive for CG7870 replication, but able to amplify wild-type or recombinant viruses. Numerous tumor and normal cell lines were evaluated for their ability to support CG7870 replication by comparing their yields of CG7870 to the yield of LNCaP cells, a prostate cancer cell line that is permissive for CG7870 replication. Virus yields were evaluated 3 days after infection at an MOI of 10 vp/cell (Fig. 1). PC-3 cells, a prostate cell line, had a virus yield similar to that of LNCaP cells, which produced on average 4.5 × 107 pfu/ml. The virus yields of other tumor cell lines (Hep G2, LOVO, MCF-7, and OVCAR-3) were about two to three logs lower than that of LNCaP. All four normal cell lines (ARPE, WI-38, IMR-90, and primary lung fibroblast cells) had a significantly reduced virus yield, especially the two lung fibroblast cell lines WI-38 and IMR-90, which gave four to five logs lower virus yield than did LNCaP cells.
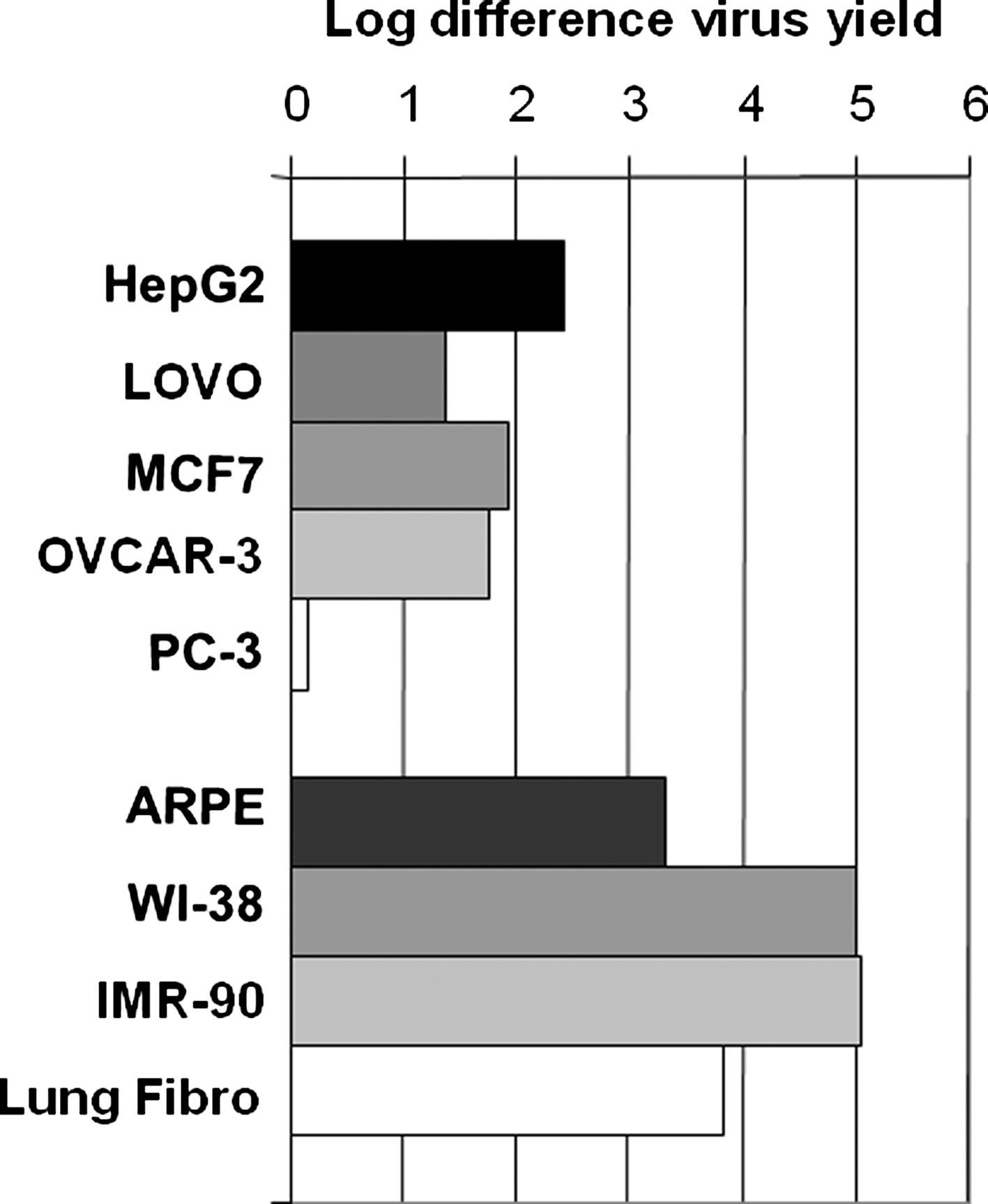
CG7870 virus yields in tumor and normal cells. The virus yield of CG7870 3 days after infection was determined for the indicated cells and LNCaP cells. The difference in the CG7870 yield between each tested cell line and LNCaP cells is shown in log scale. The average yield from LNCaP cells was 4.5 × 107 pfu/ml.
The two normal lung fibroblast cell lines were further evaluated for their ability to support CG7870 replication using MOIs from 1 to 3,000 (Fig. 2A). When the MOI was 100 or less, the two cell lines had far lower virus yields (four to five logs) than did LNCaP cells, but the difference decreased, dropping to two to three logs, when the MOI was greater than 1,000 (Fig. 2A). WI-38 cells were also tested for their permissiveness for wild-type adenovirus replication. Virus yields in WI-38 of both CG7870 and a second oncolytic adenovirus, CG8840 (which is selective for replication in bladder cells [Table 1]), were compared with that of the wild-type Ad5 over a wide range of MOI (Fig. 2B). Ad5 had a virus yield similar to CG7870 and CG8840, but at an MOI value four logs lower, demonstrating that WI-38 cells were significantly more permissive to Ad5 than to oncolytic adenovirus, as was expected. Therefore, WI-38 cells were selected for further development in a bioamplification assay.
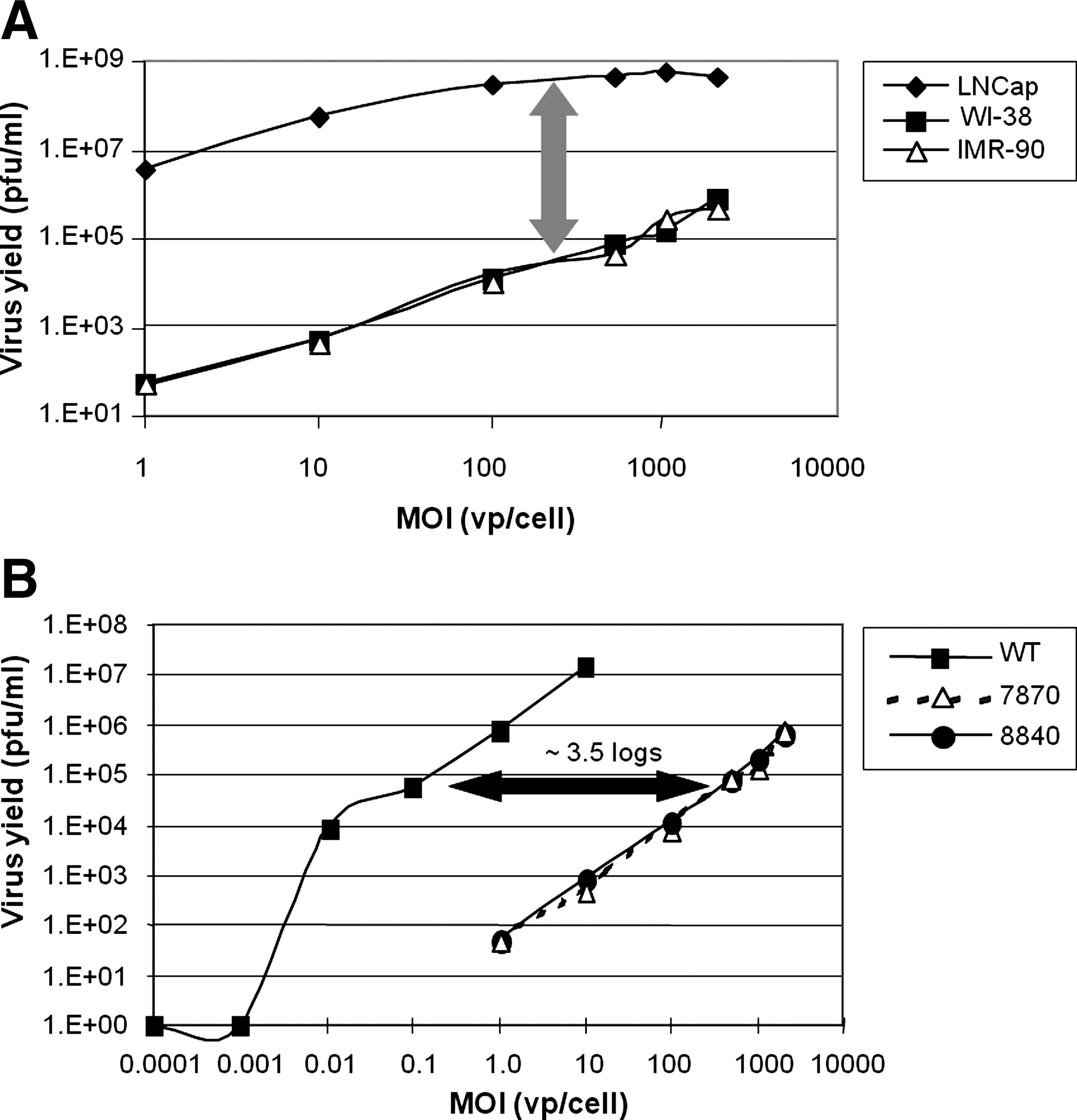
Virus yields in LNCaP, WI-38, and IMR-90 cells.
Development work leading to final assay design
Once the amplification cell line was selected, extensive work was done to establish the assay format. In the first stage, infection conditions such as the cell culture device, cell number, culture medium volume, MOI, and infection time were explored using a reference lot of CG7870 that was negative for wild type by the Ad5 LA-qPCR. The results showed that T-300 culture flasks provided the largest number of cells in the smallest volume of culture medium during infection, thus allowing better virus infection (data not shown). To further optimize infection conditions, flasks were immobilized for the first 24 hr after infection and placed on a rocking platform for an additional 24 hr. This stringent immobilization/rocking protocol was found to be important for consistent detection of the spiked Ad5 (data not shown). Additionally, the inoculate must be removed after this infection period to prevent interference with the analysis of the progeny virus population, which was allowed to amplify for another 5 days.
In the second stage of development, the number of replicates and the passage number required to achieve a high probability of detection were carefully studied. It was found that 106 CG7870 particles spiked with one Ad5 particle consistently produced a significant increase in the wild-type frequency in the range of 1:103 to 1:104 after one passage in WI-38 cells. Therefore, two replicates were selected for spiked samples with an Ad5 frequency at 1:106. For the samples spiked with Ad5 at 1:107 particles, only some replicates showed an increase in wild-type frequency after one passage, but all replicates showed an increase after two passages. To ensure that a detectable infection with Ad5 would occur in the first passage, the replicate number was determined to be four for the 1:107 spike. For the standard with Ad5 at 1:108 particles, the number of replicates showing a detectable increase in wild-type frequency varied from one quarter to three quarters of the replicates tested after three passages. As most of the production samples to be tested were expected to have a similar or lower wild-type frequency, the choice of the replicate number for this spike level was the most important one. Not only was there the consideration that bioassays are highly variable, but also a conservative assumption that only a quarter of the replicates would give positive results. Thus, it was estimated that with 20 replicates for each test sample, about five positive replicates could be observed at the end of each assay.
The final stage of the assay development was selection of the endpoint assays for the amplified virus preparation. Cell lysate from each flask was subjected to a DNA isolation step and tested by the CG7870-specific and wild-type Ad5 LA-qPCR assays to monitor the yield of both populations. A sample was considered to have an increase in the Ad5 population when it gave at least 500 copies per reaction in the LA-qPCR assay, a cutoff that was chosen based on the sensitivity of this LA-qPCR assay in a background of residual HEK293 DNA present in these crude samples and assay development experience that had shown amplification was many logs higher. Additionally, amplified materials were tested for their selectivity in killing prostate and normal cells (potency), and for genetic changes by restriction enzyme digestion of purified virus population.
Assay qualification for CG7870
Four qualification assays were performed to validate the assay design and to ensure that the sensitivity of this assay was suitable for testing production samples. The same reference standard, a CG7870 process development lot that was RCA-negative by LA-qPCR, was used in all four qualification runs, and was intended to be run as a negative control. In each assay, the reference standard was also spiked with Ad5: the 1:108 spike had 20 replicates; the 1:107 spike had four replicates; and the 1:106 spike had two replicates. Each run had one test sample, which was one of four lots (A–D) of CG7870 that had good safety profiles in patients in two early clinical trials (Husak et al., 2003; Li et al., 2004; Small et al., 2006). The number of replicates for each test sample was conservatively estimated based on the Ad5 frequency determined by the LA-qPCR, because the correlation between the PCR result and the biological frequency had yet to be established. For example, lots with an Ad5 frequency of 1:107 were tested in 20 replicates. Two replicates of each of the test samples were spiked with 1:106 Ad5 as controls to ensure that it was detectable in the production lots. All qualification runs were performed with four passages. It should be noted that in the first two runs (in which lots A and B were tested), the unspiked CG7870 reference standard was tested in two replicates; however, in these assays, wild-type–like adenovirus activity was detected even though the samples had been negative for wild-type sequences by the LA-qPCR. Therefore, in the next two qualification runs, the unspiked replicates for the reference standard were increased to 20. The design of these four qualification runs is summarized in Table 2, and a schematic of the assay is presented in Fig. 3A.
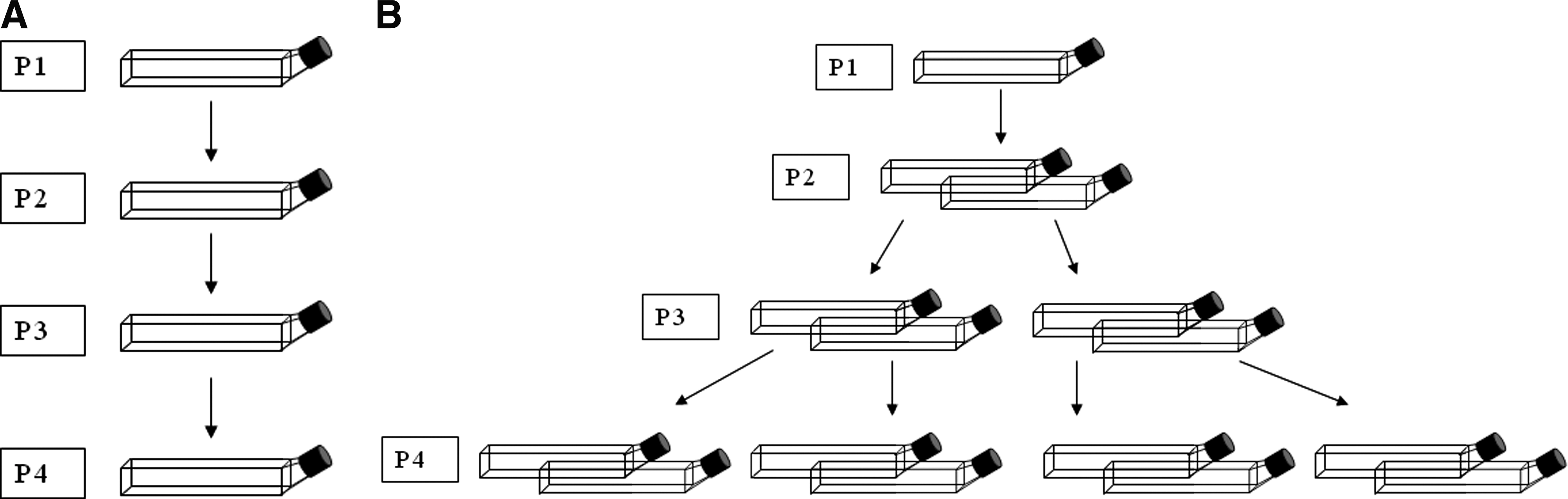
Passaging of replicates in the bioamplification assays.
Figure 4 presents LA-qPCR reference standard data from all four passages of the third qualification run. The amplification of the Ad5 over the four passages is readily apparent. Most of the passage 1 materials had fewer than 500 copies of Ad5; however, by passage 2, there begins to be a clear increase in copy numbers, with a significant increase of up to 108 wild-type copies by passages 3 and 4. Table 3 summarizes the data for the reference standards from all four qualification runs. The average percentage of wild-type–positive flasks in the second passage was 100% for the 1:106 spike, 56% for the 1:107 spike, 28% for the 1:108 spike, and 11% for the unspiked material. The number of positive flasks stabilized by the third passage (100% positive for the 1:107 spike, 63% for 1:108, and 30% for the unspiked). These data indicate that not only is the assay able to detect wild-type recombinants in a background of 3 × 109 particles, but it is sensitive enough to detect wild-type particles in a sample that was negative for wild-type by LA-qPCR.
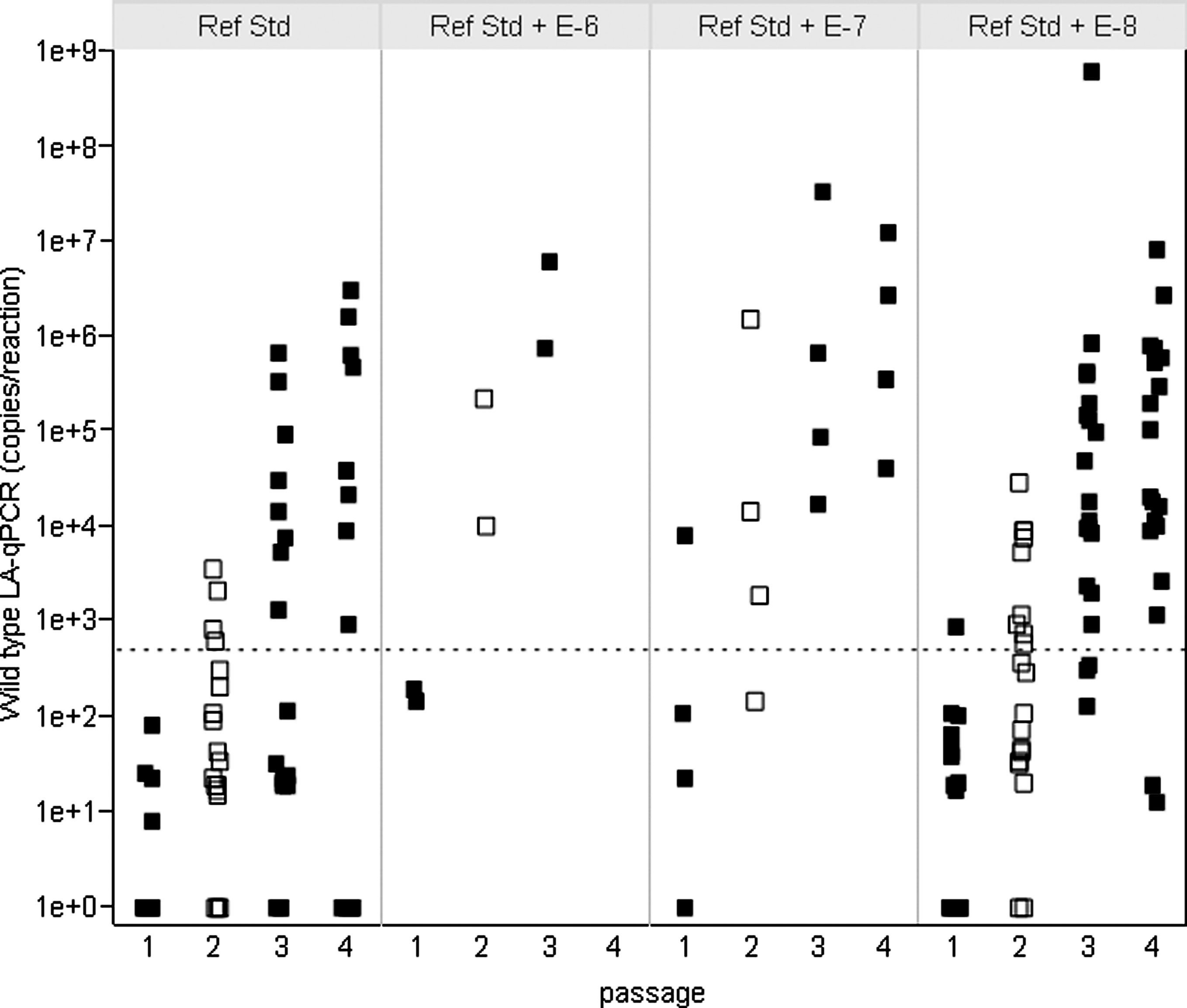
Amplification results for CG7870 reference standards. The plot represents all of the LA-qPCR results for each of the amplified CG7870 reference standards from qualification run 3. Because of the log scale used in this figure, samples that were negative (0) by LA-qPCR were converted to 1 in order to be shown. Each filled square (passages 1, 3, and 4) or open square (passage 2) indicates the result from a culture flask in each passage. The dotted line indicates 500 copies per reaction.
The number of flasks showing increased wild-type–like recombinant yield per the total number of flasks in each passage is indicated. The average positive replicate rate is indicated in parentheses.
This control lot was tested with only two flasks per passage because it was used as the negative control. After it demonstrated wild-type–like biological activity, it was tested as a regular sample with 20 flasks per passage in the last two experiments.
Flasks lost to contamination.
These positive flasks had a detected yield of wild-type–like virus just at the detection limit of the qPCR assay specific for the E1A promoter of Ad5. This may reflect the variation of the qPCR assay instead of a true increase in wild-type–like virus yield. Therefore, the percentage of positive replicates could not be accurately calculated.
Poisson distribution was used to calculate the assay sensitivity because every replicate flask represented an independent event. In the bioamplification assay, one unit of detectable analyte is defined as the detectable biological activity of the wild-type viruses or wild-type–like recombinants. Using this approach, the positive replicates from the second passage of the CG7870 bioamplification assay (Table 3) were used to calculate the detectable units per replicate for each spike level using the calculations described in Materials and Methods. The 1:108 spike had 0.32 unit per replicate, and the unspiked sample had 0.12 unit per replicate. Therefore, the difference of 0.2 unit between the 1:108 spiked and unspiked samples should represent the amount of wild-type adenovirus spiked per replicate, which was 30 vp. Poisson distribution predicts that in any assay with 20 replicates, with each replicate containing an average of 0.2 unit of detectable analyte, there would be a 98% probability of detecting at least one positive result. Therefore, the sensitivity of the assay, when performed for two passages and 20 replicates, was 30 vp of Ad5 per 3 × 109 vp of CG7870. This sensitivity was slightly better than the limit of detection in the LA-qPCR assay, which was 50 vp of Ad5 per 2.5 × 109 vp of purified CG7870, and much better than the limit of quantification, which was 500 vp per 2.5 × 109 vp of CG7870 (data not shown).
Figure 5 summarizes the Ad5 copies detected in amplified materials from lots A–D. As lot A had a high frequency of wild-type–like recombinants (1 in 4 × 105) as determined by the LA-qPCR and the CG7870-specific qPCR assays, it was tested with eight replicates per passage, whereas other lots were tested in 20 replicates per passage. After two passages, lots A and B replicates were 100% positive for wild-type–like virus yield, and lots C and D were 65 and 70% positive, respectively (Fig. 5A). The 1:106 spikes were all positive (Fig. 5B). Their approximate levels of biologically active wild-type or wild-type–like recombinants calculated by Poisson distribution analysis were comparable to that determined using the LA-qPCR assay (Table 4).
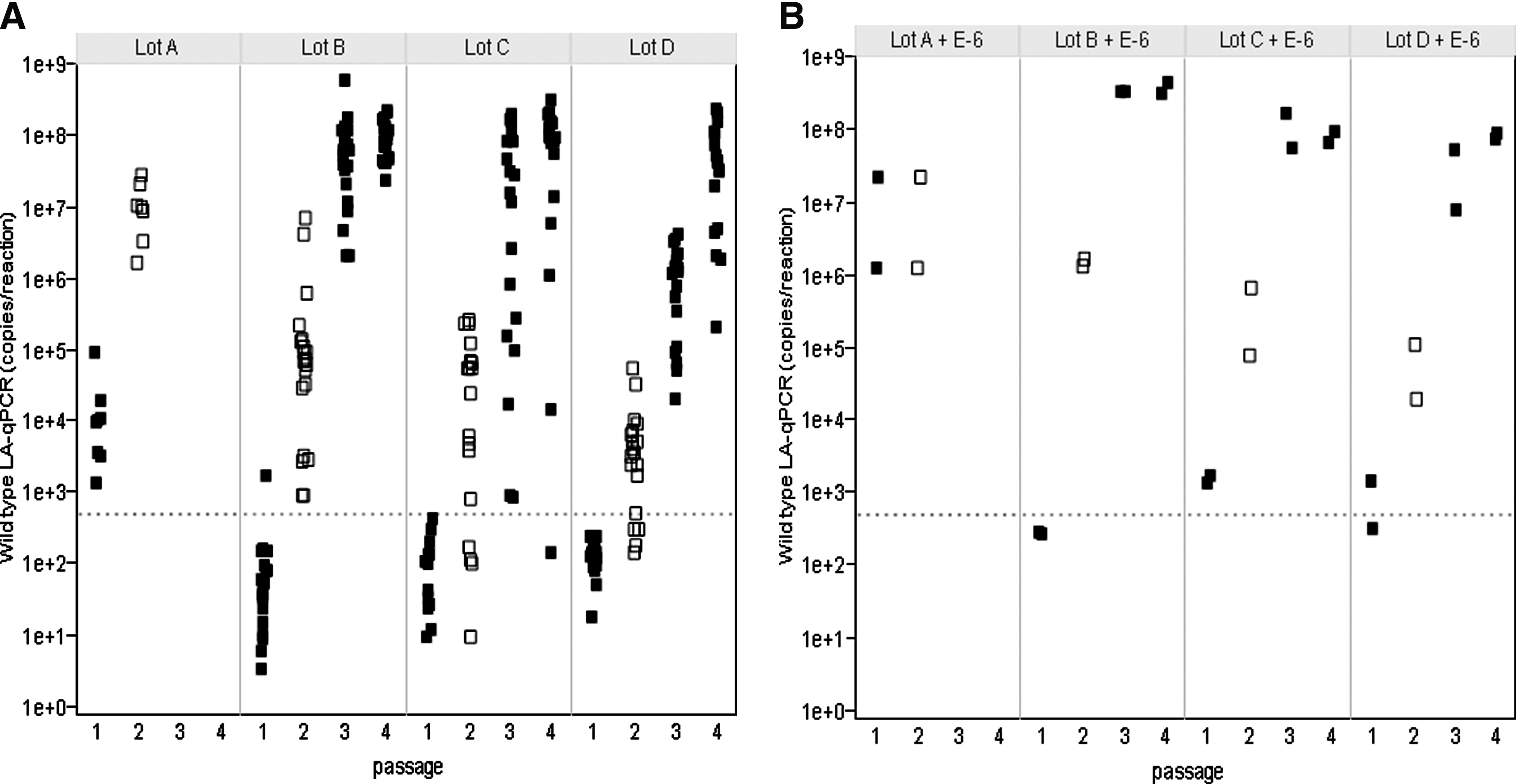
Amplification results for lots A–D and the spike controls. All of the LA-qPCR results are shown for
The amplified wild-type–like virus from the four lots was further characterized for biological function with potency assays. The four lots and the CG7870 and Ad5 controls were incubated on WI-38 cells. On day 7, the cells were exposed to alamarBlue, where higher signal levels represent greater metabolic activity. The curve of alamarBlue was subjected to four-parameter curve analysis, and the EC50 was used as the surrogate for the potency of killing these cells. The amplified virus demonstrated stronger potencies in killing WI-38 cells after two to four passages than did the starting materials (Table 5). Of the four lots, lot A had the highest wild-type–like recombinant frequency and was only amplified for two passages. All of the lot A passage 2 (P2) virus was as potent as Ad5 in WI-38 cells; the EC50 decreased significantly from 40,665 in the starting material (P0) to 758 for P2, which is similar to the EC50 of 1,122 for Ad5 in the same potency assay. Similar results were seen with the P4 results of lots B–D: the EC50 had results comparable to those of Ad5. This increase in killing potency likely reflects the much higher proportion of wild-type–like virus in the passaged samples.
The EC50 values for wild-type Ad5 and CG7870 controls were based on the viral particles per cells; the EC50 values for the passages were from the PCR-quantitated genomic copies of Ad5 + CG7870. These controls were tested in the same assays as these passage materials.
Three sets of restriction enzyme digests were performed on the amplified virus from lots A–D to look for genetic changes. The enzymes were selected for regions that would highlight differences in banding patterns between unamplified CG7870 and Ad5, which were digested for comparison. The amplified virus from each of eight flasks from the second passage of lot A showed a restriction digest pattern containing bands characteristic of both Ad5 and CG7870, as well as several unidentifiable bands that were not further characterized. Two of the eight replicates are shown in Fig. 6A. In lots B–D, all progeny virus had genetic fragments characteristic of both CG7870 and Ad5, but no extraneous bands were seen (lot B shown in Fig. 6B; lots C and D, data not shown).
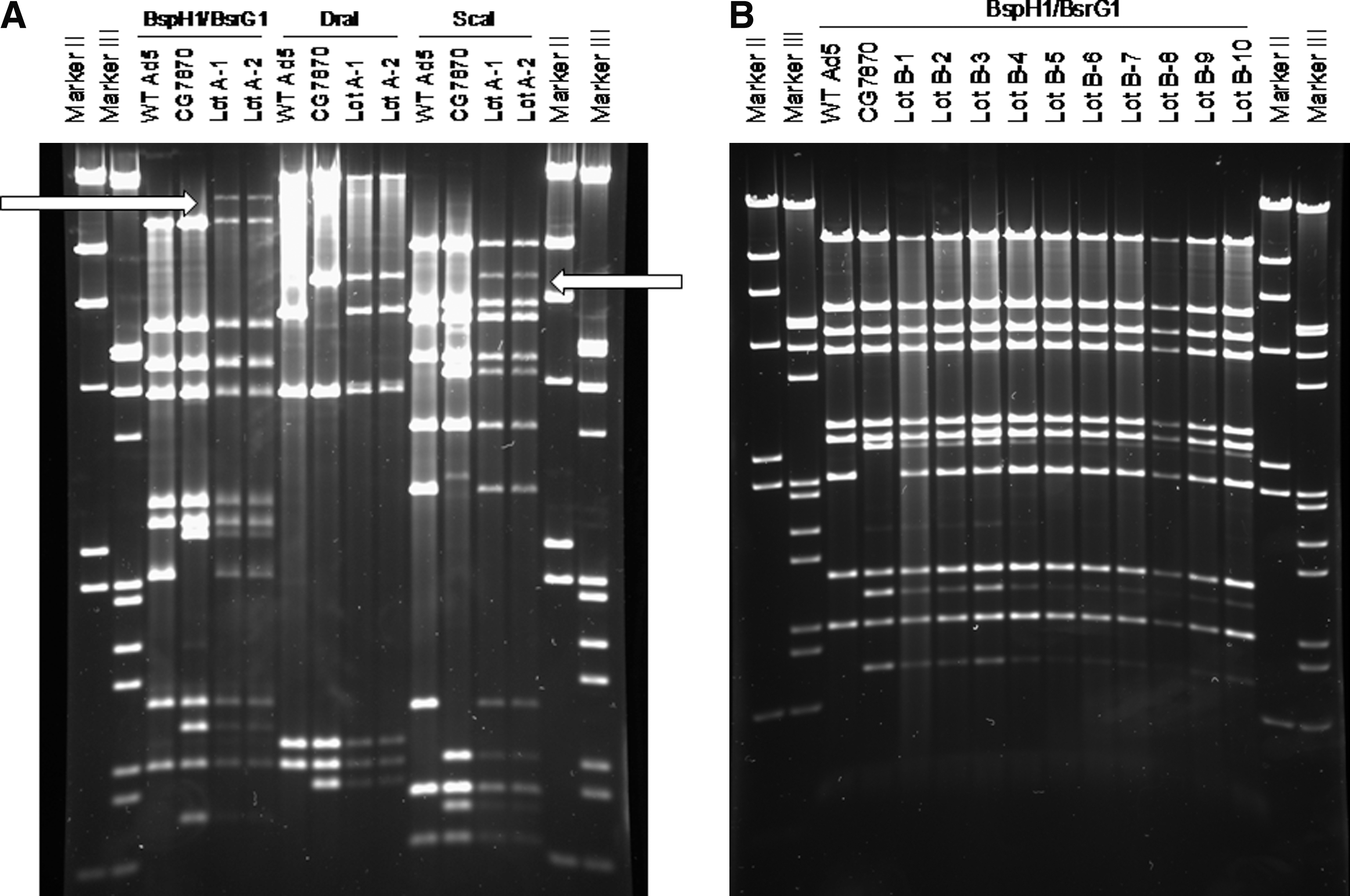
Genetic mapping of CG7870 lots A and B after bioamplification.
Bioamplification results of CG0070
Although CG0070 was negative for Ad5 E1A promoter sequences in the E1A-specific qPCR assay (data not shown), it was further tested using a modification of the CG7870 bioamplification assay to ensure that it did not contain any nonselective recombinants. To evaluate the genetic stability of CG0070, it was passaged on both MRC-5 and WI-38 cells, because the two different cell lines would potentially apply different selective pressure and thus differentially trigger genetic recombination. It should be noted, however, that such an occurrence is unlikely under the typical conditions for production of oncolytic adenoviruses, because they are produced in permissive cell lines. To further increase the likelihood of capturing any emerging recombinant, the assay was modified to passage more of each amplified material in each subsequent passage. MRC-5 and WI-38 cell cultures were infected with CG0070 using five flasks per cell line in the first passage. In passage 2 (P2), cell lysate containing amplified virus from each flask of the first passage (P1) was used to infect two flasks of cells such that P2 consisted of 10 flasks. This was repeated for a total of four passages, resulting in a total of 40 flasks (Fig. 3B). As CG0070 did not contain any wild-type sequence in the starting material, a qPCR specific for wild-type E1A promoter sequence could not be used to monitor the increase of wild-type–like recombinants. Instead, the total adenovirus yield for all flasks from each passage was determined using a commercial qPCR assay (Adeno-X qPCR assay from Clontech, Mountain View, CA) that can detect all adenovirus constructs, including CG0070 and all potential recombinants. Virus yields did not increase with passaging; they remained in the range of 1–5 × 109 adenovirus genome copies/ml between the first passage (P1) and the last (P4) for both cell lines.
CG0070 potency in WI-38 and MRC-5 cells also did not change between the starting material and the fourth passage material; the cell kill curves were essentially equivalent. Because the passaged materials did not completely kill the WI-38 or MRC5 cells, it was not possible to calculate the EC50 values. However, by extrapolating the curves to a complete kill, the EC50 values for P0 and P4 amplified in either cell line could be estimated to be 500,000 or higher (data not shown). The consistent yield and potency values of P0 and P4 materials in normal cells indicate that the amplified CG0070 did not contain highly replicating recombinants that could cause a significant increase in progeny virus in later passages. However, these results could not rule out the possibility that very small amounts of other recombinants may have emerged during bioamplification, so a subsequent restriction enzyme digestion study was conducted.
Genetic analysis of the DNA extracted from the amplified CG0070 (P4) using restriction enzyme digestion with two enzymes, BsrGI and XhoI, showed that all 40 postamplification flasks of CG0070 from each cell line had restriction maps identical to the map of the starting CG0070; a subset of representative samples from both cell lines is shown in Fig. 7. The frequency of a genetically distinct recombinant would have to be at least 5% (data not shown) for the amplified vector to exhibit a different restriction enzyme digestion pattern than the starting material, and a more sensitive method is needed to detect a lower frequency of recombinants.

Genetic mapping of CG0070 after bioamplification. The adenoviral DNA extracted from P4 of CG0070 amplified in WI-38 cells
To verify that the CG0070 materials were free of genetic recombinants that had either lost tumor selectivity or the GM-CSF expression cassette, DNA extracted from the P4 samples was subjected to three PCR reactions specific for critical regions of the CG0070 genome as described in Materials and Methods: the E2F-1 promoter region, the 5’ and 3’ junctions of the inserted human GM-CSF gene and the E3 region of the Ad5 genome. All PCR products exhibited only a single band that matched the corresponding PCR product from the unamplified CG0070 after agarose gel electrophoresis. Data from the P4 DNA amplified in WI-38 cells is shown in Fig. 8 (MRC-5 data not shown). The results of these three characterizations suggest that no genetic recombinants were amplified or emerged during serial passages of CG0070 in MRC-5 and WI-38 cells.

Verification of critical genetic elements of CG0070 after bioamplification. The adenoviral DNA extracted from P4 of CG0070 amplified in WI-38 cells was subjected to three PCR reactions.
Discussion
A bioamplification assay that serially passages oncolytic adenovirus in nonpermissive cells to amplify nonselective recombinants has been developed to measure the biological activity of potential wild-type–like recombinants and to determine the genetic stability of conditionally replication-competent oncolytic adenoviruses. The assay demonstrated great sensitivity in detecting small amounts of spiked wild-type adenovirus in the virus preparations, providing results consistent with PCR-based assays. When the assay was used to assess the genetic stability of CG0070, which contained no PCR-detectable wild-type–like recombinants in the starting material, it provided a thorough evaluation of the probability of generating recombinants when CG0070 was subjected to a high selection pressure.
During assay development, it became apparent that one passage on WI-38 cells did not comprise a sufficiently sensitive assay to detect the low level of wild-type–like recombinants that were typically found in CG7870 lots (approximately one wild-type–like recombinant genome in 1 × 106 to 1 × 107 CG7870 genome copies) when tested using qPCR assays. Therefore, the development effort focused on determining both how many cycles of amplification and how many replicates in each cycle were required to produce an assay of sufficient sensitivity, as well as the optimal procedure for virus infection of the cells. Four passages were used as a compromise between the goal of increasing the chance of detecting the changes in potency and genetic mapping, and the need to keep the assay manageable. Based on our development experience, the number of replicates per passage could be reduced when the tested sample contained a high level of wild-type–like recombinants detected by LA-qPCR (higher than 1 in 107). Nevertheless, 20 replicates were chosen for the final assay, because it is suitable for most samples which contain either low frequency or no known amounts of recombinants. In the final format, a typical bioamplification assay tested 20 replicate samples of CG7870, each using 3 × 109 vp to infect WI-38 cells in a T-300 culture flask, for a total of 6 × 1010 vp of CG7870 tested per assay, representing about 0.1% of the lot.
Although this assay format was suitable for both CG7870 and CG0070, its suitability for use with other oncolytic adenoviruses, including the specific nonpermissive cell lines used, the number of passages, and the precise method of analytical analysis of the amplified progeny virus must be determined for each specific virus construct. First, the bioamplification assay relies upon the permissiveness of the amplifying cells for nonselective recombinants, meaning that only these recombinants, and not the oncolytic adenovirus, will show exponential growth with passage. In other words, recombinants containing wild-type E1 promoter can be expected to replicate in WI-38 and MRC-5 cells at a significantly higher rate than would the conditionally replication-competent oncolytic adenovirus vectors, which are designed not to replicate in normal cells. The relative permissiveness of WI-38 or MRC-5 cells for each specific oncolytic adenovirus (and their respective potential recombinants) may be different, so it is unlikely these two cell lines can serve as universal “nonpermissive” cell lines for all oncolytic adenoviruses. In our laboratories, WI-38 cells appeared to be suitable for all the oncolytic adenoviruses that were tested, including CG7870, CG8840, and CG0070. In addition, the exact number of passages required in the assay both to provide sufficient sensitivity for the detection of existing, but very small, amounts of recombinants in an oncolytic virus preparation and to verify its genetic stability depends on two factors: (a) the magnitude of differential replication between the nonselective recombinants and the oncolytic adenovirus, and (b) the assay sensitivity needed. As the desired sensitivity for recombinants is usually in the range of 1 in 108 to 1 in 1010, one passage is rarely sufficient to detect the biological activity of the very small amounts of nonselective recombinants in the original virus preparation, even if one assumes that the recombinants replicate as efficiently as wild-type virus.
Finally, the battery of analytical methods used for studying the progeny virus population must be customized for each oncolytic adenovirus. For example, in our studies, the LA-qPCR specific to the Ad5 sequence was the key assay for detecting wild-type–like recombinants for CG7870, but was not informative in the case of CG0070 because there was no opportunity for wild-type recombination during its production. Instead, a qPCR assay that measured a generic Ad5 sequence was used to monitor the yield increase of all progeny virus, not just CG0070, in the bioamplification assay. Additionally, the selection of the restriction enzyme mapping and the potency assays for monitoring tumor selectivity and potential potency changes must be customized to each specific oncolytic adenovirus tested. To improve the chances of detecting rare or low frequency genetic recombination, it is preferable to use multiple restriction digests with different enzymes or to use PCR reactions to verify the integrity of critical genetic elements or hot spots for genetic recombinants. However, the restriction enzyme mapping can detect the presence of a different genetic species only if its abundance is greater than 5% based on spiking experiments. Thus, the absence of any extraneous band in the virus populations amplified from lots B, C, and D was not sufficient to exclude the existence of other recombinants. Although the new recombinant species detected in lot A amplified material seemed to be an interesting target for characterization, its identity was beyond the scope of this assay and was not pursued further.
The assay sensitivity was calculated from the results of samples spiked with known amounts of Ad5 into one lot of CG7870 in four experiments. Sensitivity could be calculated using the positive rates of P2 and P3 materials, because they were less than 100% positive (the Poisson calculation cannot be used when the positive rate is 100%). With CG7870, the P2 data were used for the assay sensitivity calculation, because the four cGMP lots of CG7870 either were only passaged twice (lot A) or had 100% positive rate at P2 (lot B) or at P3 (lot D). With CG0070, spiking with Ad5 could not provide a meaningful surrogate, because CG0070 did not contain recombinants with wild-type sequence. Because of this limitation, the assay sensitivity for assessing genetic stability could not be accurately estimated. Nonetheless, the authors would recommend that any new oncolytic adenovirus preparation being tested be passaged four times to ensure sufficient amplification of any nonselective recombinants to a detectable level: two to three passages appear to be enough to amplify small amounts of recombinants, and the fourth would allow for confirmation of the results.
As this bioamplification was performed using nonpermissive cell lines, it was possible this assay applied a sufficiently high selection pressure to cause mutation of the oncolytic adenovirus so that the vector harvested at the last passage might contain new recombinants not present in the starting material. This concern appears reasonable, because selection pressure has been used to promote genetic recombination to generate mutants with desired properties (Yan et al., 2003; Gros et al., 2008). However, these experiments commonly use 20 passages to achieve the desired degree of recombination (Yan et al., 2003) compared with the four passages in this assay. As was expected, there was no evidence of recombination when CG0070 underwent four passages.
As an interesting side note, the results from the bioamplification assay also support the suggestion that production of an adenovirus in HEK293 cells has a higher likelihood of generating wild-type–like recombinants, because the viral Ad5 E1 gene is present in these cells. Lot A of CG7870 was produced in HEK293 cells from the master virus bank through the final product and had the highest level of wild-type–like recombinants detected by both LA-qPCR and bioamplification assays. The master virus bank for lots B, C, and D were produced in LNCaP cells with subsequent clinical lot production in HEK293 cells. These lots had lower levels of wild-type–like recombinants than lot A, probably due to the reduced number of passages in HEK293 cells. All these lots exhibited good safety profiles in patients, with no evidence of adverse effects related to wild-type adenovirus (Husak et al., 2003; Li et al., 2004; Small et al., 2006). All patients treated intravenously with CG7870 were monitored regularly for, and found free of, circulating wild-type–like recombinants, including those patients treated with lot A (Husak et al., 2003; Small et al., 2006). CG0070 was produced in the HeLa-S3 cell line, which does not contain the viral Ad5 E1 gene, was never passaged in HEK293 cells, and was free of wild-type–like recombinants. A qPCR assay specific for wild-type E1A promoter was also used to monitor for circulating Ad5 or wild-type–like recombinants in CG0070-treated patients, with no evidence of wild-type virus detected (data not shown).
Although the bioamplification assay can detect the biological activity of contaminating recombinants in oncolytic adenovirus preparations, even when the molecular structures of these recombinants have not been identified, the assay is probably not practical for the routine testing of every production lot as a release assay due to its labor- and time-intensive nature. The value of this assay is in helping design and/or select meaningful lot release assays for new oncolytic adenoviruses. When purified lots of an oncolytic adenovirus contain PCR-detectable recombinants, this bioamplification assay could be used to verify whether the detected molecular entity has any undesired biological activity. If the answer is yes, and the results are consistent with PCR results, such as for CG7870, the PCR assay may serve as a surrogate for detecting the undesired recombinants and a specification for lot release may be justified. One could argue that this bioamplification assay would need to be used to test all lots if the PCR and bioamplification results did not correlate. There were no data to support or oppose this argument, because the PCR results were consistent with the bioamplification assay for the limited number of oncolytic viruses examined in these studies. When all lots of an oncolytic adenovirus have no PCR-detectable recombinants, as for CG0070, this assay could help assure that there are no low frequency recombinants emerging during production processes. If this assay were to detect recombinants, a relevant PCR assay could be designed based on characterization of the recombinants and used as a lot release assay. If no recombinants are detected in the bioamplification assay, a PCR assay specific for any expected genetic recombinants may be omitted from the lot release assay. However, for the safety of clinical materials, a PCR assay or assays that are specific to potential contamination, like for Ad5 or other in-house Ad5 constructs, may need to be considered.
In conclusion, this bioamplification can be conducted on each oncolytic adenovirus construct as part of a complete characterization of the adenovirus, preferably using the material generated from the final manufacturing process. This assay can both confirm the results of qPCR assays specific to known or predictable recombinants and help in the design of appropriate qPCR assays for detecting potential recombinants.
Footnotes
Acknowledgments
The authors are very grateful to Deborah Farson for providing critical review and valuable revision of the manuscript. We also appreciate Dr. Peter Working for supporting the project; Dr. D. C. Yu and Dr. Flavia Borellini for their valuable technical suggestions to this work; Ken Ho for development of the quantitative PCR assays used in the CG7870 study; and Dr. Michael Robinson for the PCR primers used in the CG0070 study.
Author Disclosure Statement
All authors performed the experiments described in this article while employed by Cell Genesys, Inc. Cell Genesys, Inc. is now BioSante Pharmaceuticals, Inc.
†
South San Francisco, CA 94080.
†
Now BioSante Pharmaceuticals, Inc., Lincolnshire, IL 60069.