
Abstract
Granulocyte colony-stimulating factor (G-CSF)-mobilized blood stem cells may become the preferable source of hematopoietic stem cells (HSCs) for gene therapy because of the higher yield of cells compared with conventional bone marrow harvesting. A G-CSF-associated risk of splenic rupture has been recognized in normal donors of HSCs, but limited information is available about the G-CSF effect in the presence of splenomegaly and extramedullary hematopoiesis. We investigated the G-CSF effect in a thalassemic mouse model (HBBth-3 ) as compared with a normal strain (C57BL/6), in terms of safety, mobilization efficacy, and distribution of stem cells among hematopoietic compartments. There was no death or clinical sequelae of splenic rupture in G-CSF-treated animals of either strain; however, hemorrhagic infarcts in the spleen were detected with low frequency in G-CSF-treated HBBth-3 mice (12.5%). HBBth-3 mice mobilized less effectively than C57BL/6 mice (Lin−Sca-1+c-Kit+ cells/μl of peripheral blood mononuclear cells [PBMCs]: 90 ± 55 vs. 255 ± 174, respectively, p = 0.01; CFU-GM/ml PBMCs: 390 ± 262 vs. 1131 ± 875, p = 0.01) because of increased splenic trapping of hematopoietic stem and progenitor cells (Lin−Sca-1+c-Kit+ cells per spleen (×105): 487 ± 35 vs. 109 ± 19.6, p = 0.01; CFU-GM per spleen (×102): 1470 ± 347 vs. 530 ± 425, p = 0.0006). Splenectomy restored the mobilization proficiency of thalassemic mice at comparable levels to normal mice and resulted in the development of a hematopoietic compensatory mechanism in the thalassemic liver that protected splenectomized mice from severe anemia. Our data imply that, in view of human gene therapy for thalassemia, either multiple cycles or alternative ways of mobilization may be required for a sufficient yield of transplantable HSCs. In addition, strategies to minimize the risk of G-CSF-induced splenic infarcts should be explored in a clinical setting.
Introduction
Autologous hematopoietic stem cell (HSC) harvest in thalassemia has been largely unexplored, as gene therapy has not long been considered to have realistic therapeutic potential for the disease. For effective gene therapy, there is a need for high numbers of transduced hematopoietic stem cells. G-CSF-mobilized peripheral blood stem cells (mPBSCs) provide a 3- to 4-fold higher CD34+ cell yield than conventional BM harvesting under a less invasive procedure and may also allow higher rates of gene transfer than similar cells from steady state BM (Dunbar et al., 1996; An et al., 2001; Horn et al., 2004), although this is still controversial (Thomasson et al., 2003). Thalassemia lacks a selective survival advantage at the stem cell level, and thus a high number of transduced HSCs will be required to effectively compete for niche over endogenous BM cells. Therefore, PBSCs may become the preferable source of SCs in clinical applications of thalassemia gene therapy.
However, there are some specific clinical features in thalassemia that could potentially affect mobilization. Extramedullary hematopoiesis could result in sequestration of HSCs in the spleen during mobilization, thus effectively reducing the yield of CD34+ cells. Moreover, preexisting splenic enlargement may increase the rare but existing risk of splenic rupture after G-CSF treatment. On the other hand, a chronic hypercoagulable state has been recognized in thalassemia (Eldor et al., 1999; Eldor and Rachmilewitz, 2002), which in combination with the G-CSF-induced leukocytosis and activation of white blood cells may precipitate thrombotic events.
So far, no preclinical studies on mobilization in β-thalassemia have been reported and there is only one publication on the mobilization efficacy in pediatric patients with thalassemia major (Li et al., 1999). With the goal of human gene therapy of thalassemia, the limited existing knowledge on the G-CSF effect in conditions of splenomegaly and extramedullary hematopoiesis, both in terms of safety and efficacy, has prompted us to explore G-CSF mobilization in a preclinical mouse model of thalassemia as compared with the normal C57BL/6 strain.
Materials and Methods
Mice
Mice were maintained in a conventional clean facility in accordance with the local institutional animal care and use committee. The thalassemic mouse model for β-thalassemia (HBBth-3 /HBBth-3 ), developed by Yang and colleagues (1995), was used in the experiments (Jackson Laboratory, Bar Harbor, ME) at 8–12 weeks of age. The heterozygous HBBth-3 animals represent a viable form of the disease, which clinically resembles human β-thalassemia intermedia. Their response to G-CSF priming was compared with that of age-matched C57BL/6J mice.
Mobilization
Recombinant human G-CSF (huG-CSF; Amgen, Thousand Oaks, CA) was administered intraperitoneally at 200 μg/kg in a total injection volume of 250 μl once per day for 7 days. The mice were killed 2 hr after the last G-CSF dose and tissues were collected for analysis. Control mice received no treatment.
Tissue preparation
Blood samples were collected under anesthesia by cardiac puncture in heparinized tubes. Spleen cells were prepared by mechanical dissociation with the end of a sterile plunger and gently strained through a 100-μm pore filter (Falcon; BD Biosciences, Bedford, MA). Single cells were obtained by repeatedly flushing the spleen cells through a 21-gauge needle. Bone marrow cells were harvested by flushing the two femurs with 1 ml of Iscove's modified Dulbecco's medium (IMDM)–2% fetal bovine serum (FBS) with a 25-gauge needle and filtered through a 70-μm pore filter (Falcon; BD Biosciences). Low-density peripheral blood, BM, and spleen mononuclear cells were obtained by density gradient separation (Histopaque 1083; Sigma-Aldrich, St. Louis, MO).
Splenectomy
Splenectomy was performed under general anesthesia and sterile conditions. After a small incision was made in the peritoneal wall, the spleen was removed after ligation with surgical silk of the blood vessels supporting the spleen. Mice were left to recover for 10 days before being used in the experiments.
Hematological parameters
Hematological profiles of freshly drawn EDTA-treated blood were obtained with an automatic blood cell counter giving complete blood counts. Reticulocytes were counted on peripheral blood smears after staining with 1:1 brilliant cresyl blue followed by a 15-min incubation in a water bath at 37°C.
Histopathological and immunohistochemical analysis
All morphological studies were evaluated by a pathologist. Liver and spleen were fixed after removal in 10% neutral formaldehyde buffer for 18–24 hr, dehydrated, and embedded in paraffin. Three-micron-thick sections were routinely stained with hematoxylin and eosin for histology and with Gomori histostain for reticulin fibers. Immunohistochemistry was performed with a fully automated staining system (BOND-MAX; Vision Biosystems/Leica Microsystems, Wetzlar, Germany) with an antibody panel that included the following: anti-myeloperoxidase (Ab-1, diluted 1:250; Neomarkers/Thermo Fisher Scientific, Fremont, CA), anti-c-Kit (C-19, diluted 1:100; Santa Cruz Biotechnology, Santa Cruz, CA), anti-TER-119 (Ly-76, diluted 1:150; BD Biosciences, San Jose, CA), and anti-Ki-67 (diluted 1:50; Dako, Glostrup, Denmark). Heat-induced epitope retrieval in a steamer, with citrate buffer (pH 6.0) for 20 min, was performed for all antibodies.
Hematopoietic progenitor studies
Progenitor cell analyses were carried out on three hematopoietic tissues: bone marrow, peripheral blood, and spleen. Bone marrow cells, peripheral blood mononuclear cells, and spleen single-cell suspensions were plated in duplicate at a density of 0.5 × 105, 1.0 × 105, and 0.5 × 105 cells, respectively, in plating medium consisting of IMDM, 30% FBS, 1% bovine serum albumin (BSA),
Flow cytometry
Immunostaining and flow cytometric analyses were performed according to standard procedures and cells were subsequently analyzed on a fluorescence-activated cell-sorting (FACS) flow cytometer (BD Biosciences, San Jose, CA). Briefly, for flow cytometric analysis of Lin−Sca-1+c-Kit+ cells, cells from PB, BM, and spleen were stained with APC mouse lineage cocktail (containing allophycocyanin [APC]-conjugated anti-CD3, anti-CD11b, anti-B220, anti-GR-1, and anti-erythrocyte-specific antigen) and fluorescein isothiocyanate (FITC)-conjugated anti-Sca-1 and phycoerythrin (PE)-conjugated anti-c-Kit monoclonal antibody. Lin−Sca-1+c-Kit+ (LSK) cells were defined as HSCs. Cells incubated with an isotype-matched antibody served as negative controls. Data were collected from 5 × 103 to 10 × 103 cells and analyzed with CellQuest software (BD Biosciences, San Jose, CA). The percentage of LSK cells was determined in the three hematopoietic compartments and the absolute number of LSK cells per microliter of PBMNCs, per two femurs and per spleen, was calculated on the basis of the individual mononuclear cell counts.
Statistical analysis
Differences between the different treatment groups were determined by Student t test. Values of p less than 0.05 were considered statistically significant. All results are expressed as means ± SD.
Results
G-CSF causes dramatic alterations in the histology of normal and thalassemic spleen
The spleens of untreated C57BL/6 mice showed normal parenchymal architecture with approximately equal proportions between the two pulps (Fig. 1A, panel a). Foci of extramedullary hematopoiesis were visible around the splenic cords, in the perifollicular zone, and within the red pulp. In untreated HBBth-3 spleens, increased extramedullary hematopoiesis was evident. The white pulp was almost replaced by a dilated and hypercellular red pulp with a predominance of abnormal erythrocytes trapped in the sinusoidal meshwork. This expansion of the erythrocyte-rich perifollicular zone resulted in an unclear distinction between the two pulps (Fig. 1A, panel c). Myeloid and erythroid cells lost their organoid distribution and were scattered within the reticulin meshwork of the red pulp among numerous macrophages serving as “nurse cells” and filled with hemosiderin granules (Fig. 2A, panels a, b, and c). Clusters of megakaryocytes were also visible within the red pulp (Fig. 2A, panel c). The intense hypercellularity resulted in parenchymal congestion represented by ill-defined splenic cords and fibrous septae, as shown by Gomori staining (Fig. 1B, panel b).
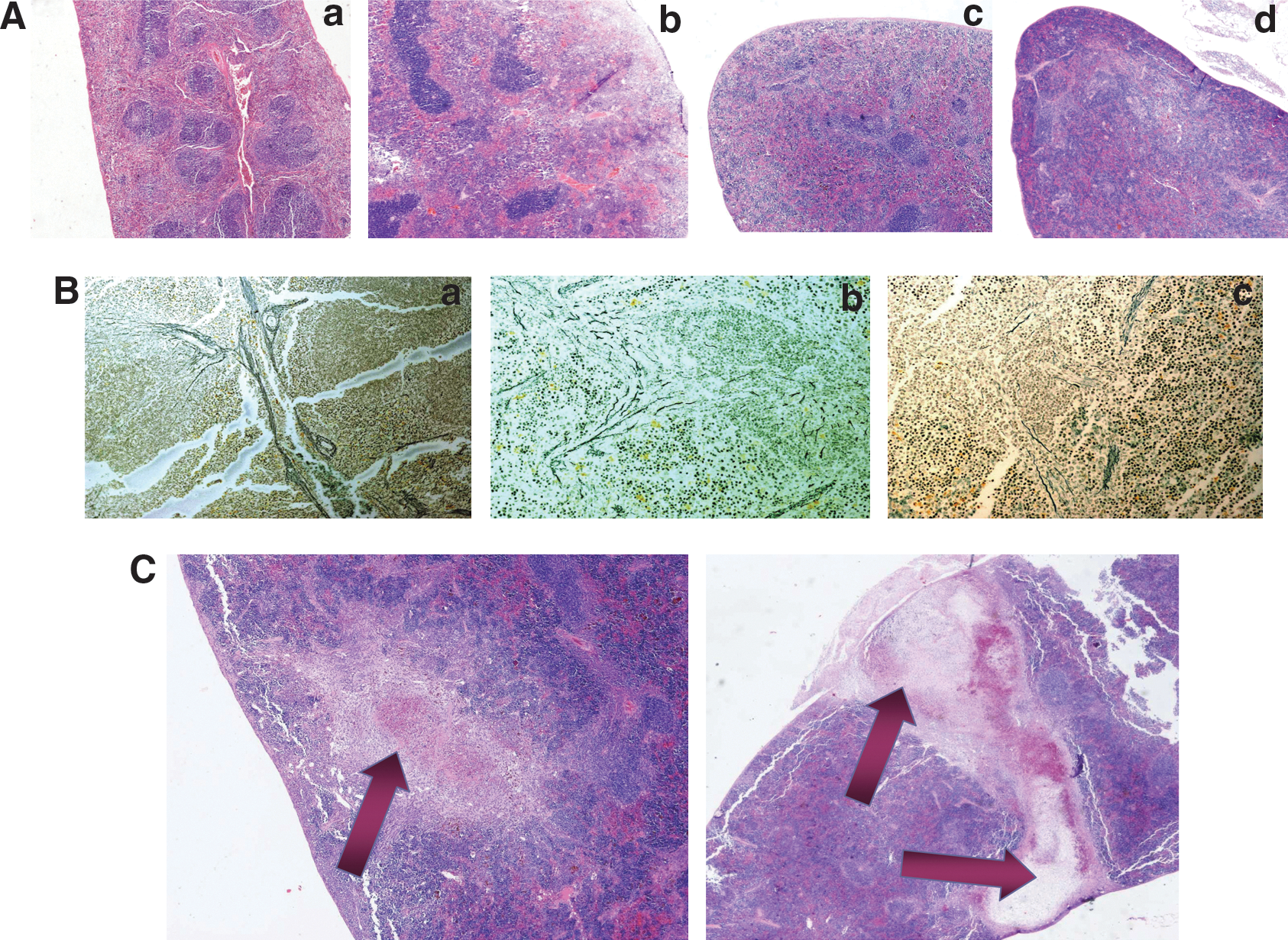
(

Extramedullary hematopoiesis in the spleens of C57BL/6 and HBBth-3
mice at steady state and after G-CSF treatment (original magnification, × 400; insets, × 600). (
After G-CSF treatment, in normal spleens there was a slight increase in white pulp in sharp contrast to an extensive dilation of red pulp (Fig. 1A, panel b). All hematopoietic lineages were represented in increased numbers with a prominent expansion of the erythroid lineage, presented with numerous erythroblasts and mature red cells in the red pulp and in the lumen of trabecular vessels (Fig. 2C, panel b). Also, a hyperplastic myeloid lineage with foci of leukocytes was arranged around splenic cords whereas clusters of myeloid precursors could be seen within the red pulp (Fig. 2B, panel b). The spleens of HBBth-3 mice, after G-CSF treatment, presented an excessively hypercellular appearance with almost absolute domination of the red over the white pulp (Fig. 1A, panel d). This was due mainly to predominant granulocytic hyperplasia with foci of neutrophils and myeloid progenitors homing around splenic cords, identified as MPO+ cells by immunohistochemistry (Fig. 2B, panel d). On the other hand, erythropoiesis in the HBBth-3 , G-CSF-treated mice, was not as strongly enhanced as myelopoiesis and the erythroid compartment was moderately expanded compared with the untreated HBBth-3 group (Fig. 2C, panels c and d). Proliferating cells (Ki-67+) in excess dominated the red pulp of thalassemic spleens, mostly in areas where cells of myeloid lineage were homing (Fig. 2D, panel d). A low degree of mitotic activity was also present in the white pulp (Fig. 2D, panel d, arrows). Gomori histostaining showed discontinued or collapsed splenic cords and fibrous septae due to excessive parenchymal congestion (Fig. 1B, panel c).
G-CSF effect on spleen and liver size
Spleen and liver weight was determined as a ratio (×1000) to total body weight. At steady state, thalassemic spleens were considerably enlarged over normal spleens (19.42 ± 5.31 vs. 4.53 ± 1.56, respectively; p < 0.0001), probably because of the increased trapping of damaged thalassemic red blood cells and the presence of intense extramedullary erythropoiesis. After G-CSF treatment, the size difference between strains was again significant (31.6 ± 7.63 vs. 10.55 ± 2.27; p < 0.0001); however, the G-CSF effect on splenic enlargement was stronger in the normal strain, finally resulting in a 133% increase (from 4.53 ± 1.56 to 10.55 ± 2.27; p < 0.0001) as compared with a 65% increase in thalassemic spleen size (from 19.42 ± 5.31 to 31.6 ± 7.63; p < 0.001) (Fig. 3A). There was no death or clinically observed splenic rupture in G-CSF-treated animals of either strain (0 of 68); however, hemorrhagic infarcts were detected in the spleens of G-CSF-treated thalassemic mice (5 of 40, 12.5%) at the time of sacrifice (Fig. 1C). Infarcts developed intraparenchymally either as hemorrhagic areas with hemosiderin deposits and debris or as well-organized lesions surrounded by dense fibrous septa (Fig. 1C). They were in all cases self-limited without causing capsular tear or/and hemoperitoneum.
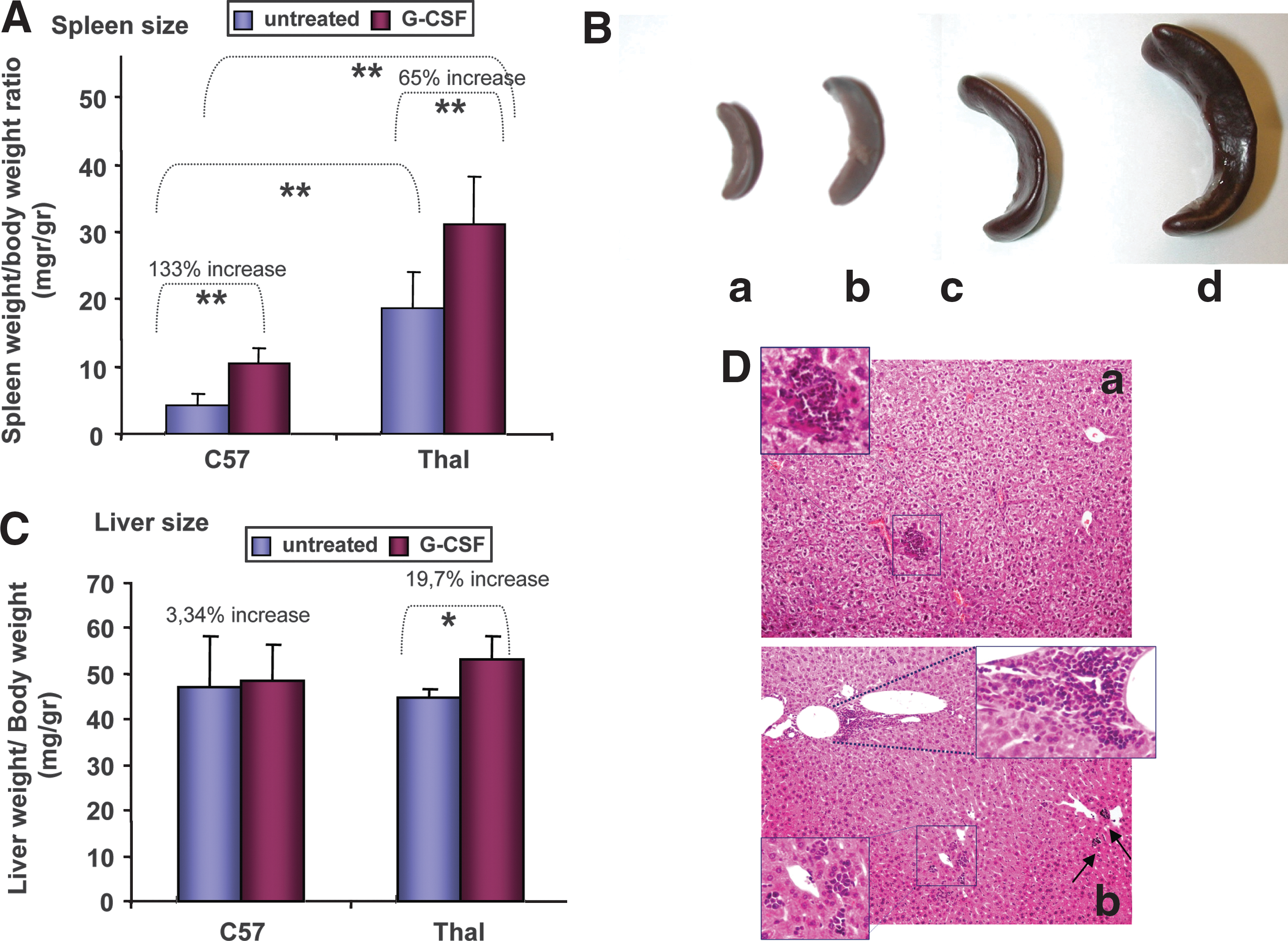
(
After G-CSF treatment, thalassemic liver increased by 19.7% (p < 0.0001) compared with the 3.3% increase (p = 0.7) in normal liver. At steady state there was no significant difference between strains (Fig. 3C). Thalassemic liver enlargement was associated with the presence of numerous erythromyeloid aggregates and scattered megakaryocytes in liver sections, suggesting either a G-CSF-induced stimulation of liver extramedullary hematopoiesis or the hepatic migration of hematopoietic progenitor cells from the circulation (Fig. 3D).
Differences between strains in mobilization and distribution of hematopoietic stem and progenitor cells in hematopoietic compartments after G-CSF
HBBth-3 and C57BL/6 mice received G-CSF for 7 days or no treatment and were killed later on day 7. Mobilization efficiency was determined by calculating the absolute numbers of LSK cells in the periphery, based on the percentage of LSK cells by flow cytometry and on the individual PBMNC counts (LSK cells per microliter of PBMNCs). Mobilization efficiency was also assessed by the numbers of circulating colony-forming cells (CFCs) in the peripheral blood, based on the frequency of CFU-GM per 105 cells plated from PB and multiplied by PBMNC count (CFU-GM per milliliter of PBMNCs). Likewise, absolute numbers of LSK cells and CFU-GM were also calculated per spleen and per two femurs by flow cytometric analysis and clonogenic assays.
Thalassemic mice yielded 2.8-fold fewer circulating hematopoietic stem cells postmobilization as compared with normal mice (LSK cells/μl PBMNCs: 90 ± 55 vs. 255 ± 174; p = 0.01) (Fig. 4A). The less efficient mobilization correlated with significantly higher accumulation of HSCs after G-CSF treatment in thalassemic compared with normal spleen (LSK cells/spleen (× 105): 487 ± 35 vs. 109 ± 19.6, respectively; p = 0.01). This was the result of a 3.8-fold increase in LSK cells in thalassemic spleens (p = 0.001) relative to the untreated state as compared with a 3-fold increase in LSK cells in normal spleens (p = 0.01) after G-CSF treatment (mean LSK cell increase/(× 105): 360 vs. 73, respectively), in addition to the 3.6-fold higher HSC content in steady state thalassemic spleen as compared with normal spleen (p = 0.01; Fig. 4A). Immunohistochemistry of spleen sections also confirmed the presence of numerous early hematopoietic progenitors (c-Kit+ cells) in the perifollicular zone and in the red pulp of thalassemic spleens with evidence of sequestration within sinusoids and trabecular vessels (Fig. 4C, panel d, magnified area). In contrast, only rare and scattered c-Kit+ cells were present in the spleens of C57 mice (Fig. 4C, panel b).
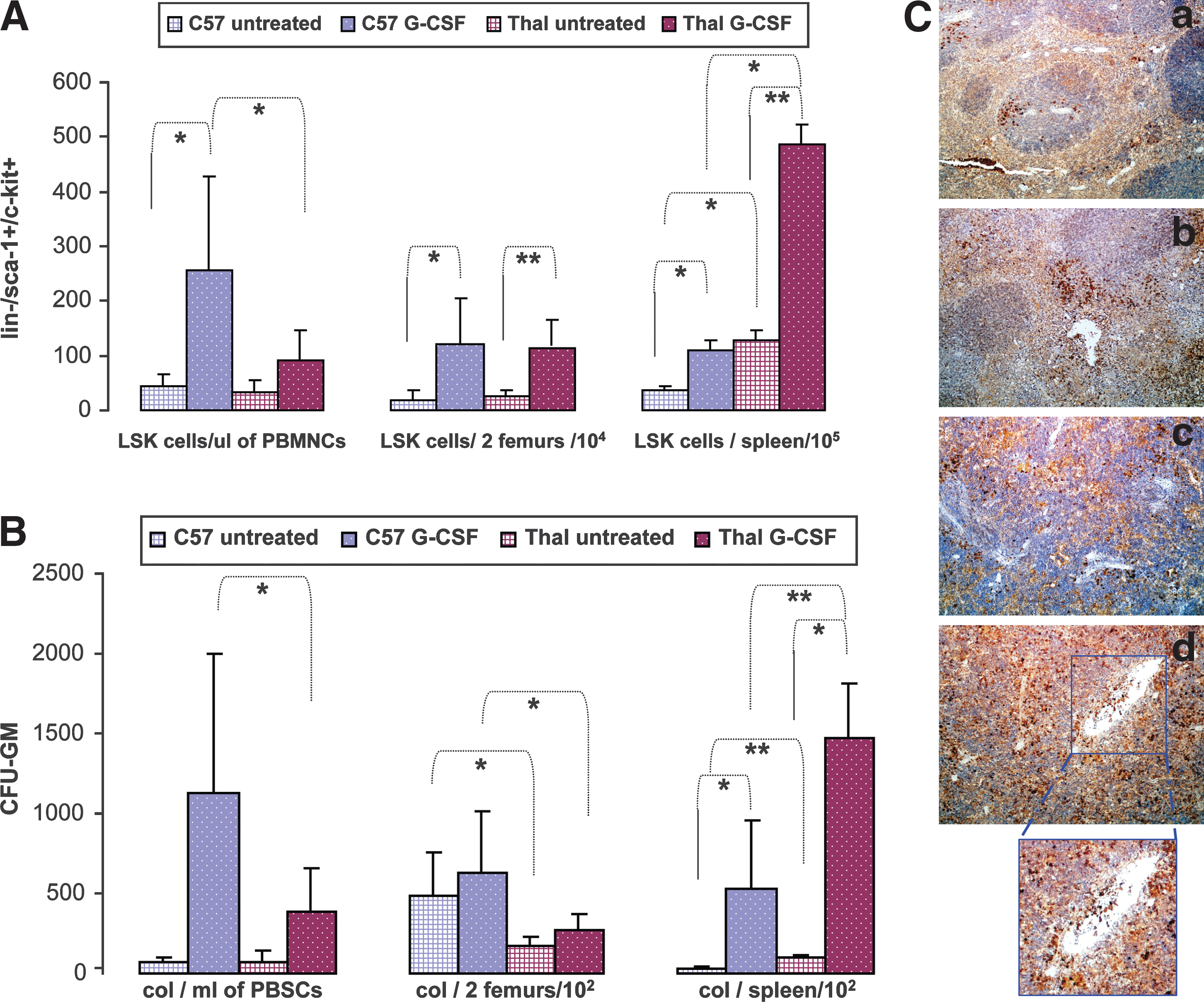
(
The BM content in LSK cells at steady state and after G-CSF treatment did not differ between HBBth-3 and normal mice (LSK cells/two femurs (104): 25.6 ± 11.5 vs. 20 ± 17.9, respectively [p = 0.5] and 113 ± 47 vs. 120 ± 85, respectively [p = 0.8]) (Fig. 4A), implying that splenic trapping was the major contributor to the deficit in circulating HSCs of thalassemic mice.
In terms of CFC mobilization, as with LSK cells also, the thalassemic strain mobilized CFCs less effectively than the C57BL/6 strain (CFU-GM/ml PBMNCs: 390 ± 262 vs. 1131 ± 875; p = 0.01) (Fig. 4B). There was again a significantly higher accumulation of CFU-GM after G-CSF treatment in thalassemic compared with normal spleen (CFU-GM/spleen (× 102): 1470 ± 347 vs. 530 ± 425; p = 0.0006) (Fig. 4B), which was associated with a higher absolute CFU-GM increase in thalassemic compared with normal spleen (mean CFU-GM increase after G-CSF/spleen (× 102): 1365 vs. 507, respectively). The total CFU-GM per spleen after G-CSF treatment increased over steady state by 23-fold in normal mice (CFU-GM/spleen (× 102): 23 ± 14 vs. 530 ± 425; p = 0.002) and 14-fold in thalassemic mice (CFU-GM/spleen (× 102): 105 ± 15 vs. 1470 ± 347; p = 0.01), as a result of the increased presence of CFU-GM in the enlarged steady state thalassemic spleen (CFU-GM/spleen (× 102): 105 ± 15 vs. 23 ± 13; p = 0.00003) (Fig. 4B).
In contrast to BM LSK cells, the BM CFU-GM content at steady state was significantly decreased in HBBth-3 mice as compared with normal mice, most likely a consequence of the expansion of the erythroid compartment within the thalassemic BM (CFU-GM/two femurs (× 102): 176 ± 59 vs. 488 ± 274; p = 0.03) (Fig. 4B). This difference between strains was also maintained after G-CSF treatment and resulted in higher numbers of CFU-GM in the primed normal BM compared with thalassemic BM (CFU-GM/two femurs (× 102): 624 ± 384 vs. 266 ± 111; p = 0.02) (Fig. 4B).
Splenectomy restores the mobilization proficiency of thalassemic mice
Splenectomy in HBBth-3 mice significantly increased the number of mobilized LSK cells compared with nonsplenectomized HBBth-3 mice (LSK cells/μl PBMNCs: 164 ± 111 vs. 90 ± 55, respectively; p = 0.04). This effect was also accompanied by a striking increase in circulating CFCs per milliliter (CFU-GM/ml PBMNCs: 2022 ± 937 vs. 390 ± 262, respectively; p = 0.0001) (Fig. 5A) after G-CSF treatment, suggesting that intrasplenic trapping is the major contributor to the inefficient mobilization of thalassemic mice. Splenectomy finally resulted in comparable mobilization between thalassemic and C57 splenectomized mice (LSK cells/μl PBMNCs: 164 ± 111 vs. 223 ± 99 respectively; p = 0.28; CFU-GM/ml PBMNCs: 2022 ± 937 vs. 1436 ± 992; p = 0.27) (Fig. 5A). Surprisingly, splenectomy did not further improve the mobilization efficiency of C57 mice (LSK cells/μl PBMNCs: 255 ± 174 vs. 223 ± 99; p = 0.57; CFU-GM/ml: 1131 ± 875 vs. 1436 ± 992; p = 0.42), suggesting that, in contrast to thalassemic mice, the major mechanism of the increased cell accumulation in primed normal spleen is the in situ G-CSF-induced stimulation of the production of stem and progenitor cells, rather than splenic sequestration (Fig. 5A).
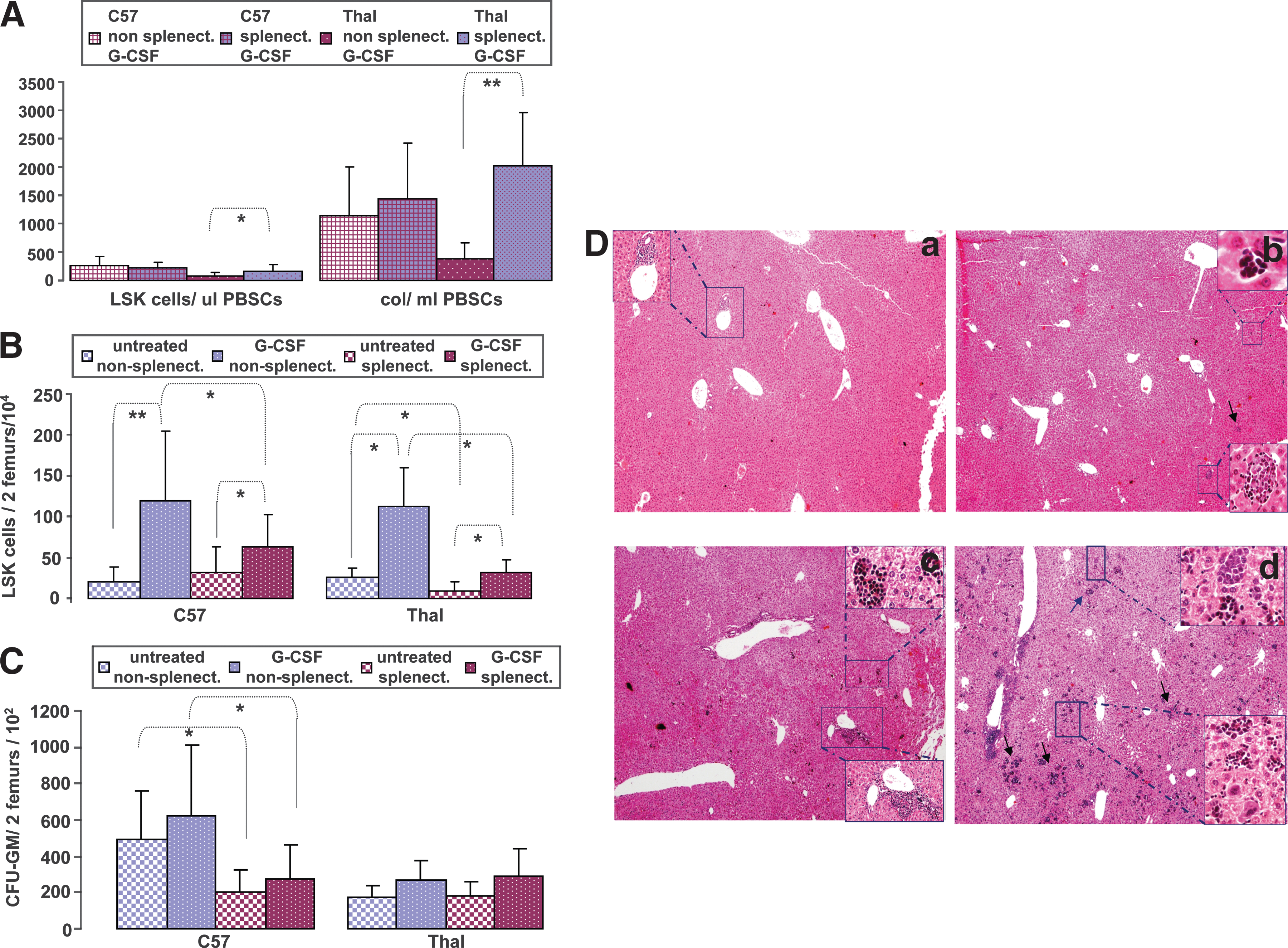
(
There was also a lower content of BM LSK cells in thalassemic G-CSF-treated splenectomized mice as compared with thalassemic G-CSF-treated nonsplenectomized mice (LSK cells/two femurs (× 104): 32 ± 15.6 vs. 113 ± 47.4; p = 0.004), which could imply an increased release of LSK cells from the primed BM after splenectomy (Fig. 5B). However, this seemed to be a consequence of the significantly decreased LSK cells in steady state BM of thalassemic splenectomized mice as compared with nonsplenectomized mice (LSK cells/two femurs (× 104): 9 ± 2.2 vs. 25.6 ± 11.5; p = 0.02, respectively) (Fig. 5B). As no difference was observed in steady state circulating LSK cells between thalassemic nonsplenectomized and splenectomized animals (data not shown), the reduced stem cell pool after splenectomy may reflect a competition effect within the BM at the level of stem cell commitment toward the erythroid lineage, because of the increased erythroid demand after removal of an active site of thalassemic hematopoiesis. The increased erythroid demand after splenectomy in normal mice resulted in significantly fewer CFU-GM progenitors in the steady state BM, as compared with nonsplenectomized mice (CFU-GM/two femurs (× 102): 204 ± 122 vs. 488 ± 273; p = 0.01), and this difference was also maintained after G-CSF treatment (CFU-GM/two femurs (× 102): 273 ± 189 vs. 624 ± 384; p = 0.02) (Fig. 5C).
Splenectomy results in the development of a hematopoietic compensatory mechanism in the liver of splenectomized thalassemic mice
Splenic hematopoiesis provides thalassemic mice with an erythropoietic compensatory mechanism, consequently resulting in the absence of severe anemia in these mice (Yang et al., 1995). After splenectomy, we observed a compensatory increase in hematopoiesis in the thalassemic liver, probably a response to increased erythropoietic stress after the removal of a major site of hematopoiesis in these animals (Fig. 5D, panel c). Liver erythropoiesis was represented by multiple intraparenchymal foci of erythroid cells and the presence of myeloid-origin cells in periportal areas. After G-CSF treatment, intense trilineage extramedullary hematopoiesis in the liver was detected in splenectomized thalassemic mice, inducing dramatic alterations in the liver histology. Confluent erythroid aggregates and numerous megakaryocytes were present in the hepatic lobules. Myeloid precursors and mature cells were arranged in well-organized colonies intraparenchymally and around the central vein or portal tracts (Fig. 5D, panel d).
In contrast, these features were rare and not as intense in splenectomized C57 animals. Untreated mice had only a minimal degree of extramedullary hematopoiesis in the liver with inconspicuous foci of hematopoietic cells, mostly erythroid, whereas after G-CSF priming, increased hepatic hematopoiesis was evident (Fig. 5D, panels a and b).
G-CSF effect on hematological parameters of HBB th-3 and C57 mice
There was a dramatic difference in hemoglobin (Hb) values and reticulocyte frequencies between HBBth-3 and normal mice (Hb: 7.21 ± 1.84 vs. 12.01 ± 2.24 g/dl, respectively; p = 0.000001 and 34.98 ± 14.67 vs. 3.62 ± 1.68%, respectively; p = 0.000001) indicative of intense hematopoietic activity in the thalassemic strain in response to the erythroid demand (Table 1). G-CSF administration did not significantly affect hemoglobin values or reticulocytes in either strain when spleens were intact. However, splenectomy in C57BL/6 mice caused anemia, as also shown by others (Cronkite et al., 1993) (Hb: 10.53 ± 1.64 vs. 12.01 ± 2.24; p = 0.00004), due to the removal of an active site of hematopoiesis and anemia worsened after G-CSF administration (Hb: 10.53 ± 1.64 vs. 9.74 ± 1.53; p = 0.004) (Table 1). In contrast, splenectomy in thalassemic mice did not significantly alter Hb values or reticulocyte frequency at steady state or after G-CSF treatment, probably due to the development of intense compensatory hematopoiesis from the thalassemic liver (Table 1).
Abbreviations: G-CSF, granulocyte colony-stimulating factor; Hb, hemoglobin.
p = 0.000001.
p = 0.00004.
p = 0.004.
Discussion
Gene therapy for thalassemia has moved to the clinic with the initiation of the first clinical trial of human gene therapy for thalassemia in France in 2006 (Bank et al., 2005). This was the reasonable consequence of several preclinical studies demonstrating correction of the thalassemic phenotype in animal models of hemoglobinopathies or in xenografts (May et al., 2000; Pawliuk et al., 2001; Imren et al., 2002; Rivella et al., 2003; Puthenveetil et al., 2004; Miccio et al., 2008) and paved the path to the initiation of clinical trials for gene therapy of thalassemia. The preferred source of HSCs to be used for genetic modification in thalassemia has not yet been determined. G-CSF-mobilized PBSCs will potentially become the target cells for gene correction over bone marrow stem cells, because of the higher yield of stem cells in the leukapheresis product (Beyer et al., 1995; Hartmann et al., 1997).
The development of animal models of hemoglobinopathies (Trudel et al., 1994; Yang et al., 1995) has offered crucial insights in the field of gene therapy. The animals heterozygous for the b1 and b2 adult globin gene deletion that we have used in our study phenotypically resemble human patients with β-thalassemia intermedia (Yang et al., 1995) and provide a naturally occurring state for studying mobilization of thalassemic HSCs. Significant interstrain variability in the ability to mobilize stem and progenitor cells, due to genetic influences, has been reported, and the C57BL/6 strain has been specifically shown to represent a G-CSF low-responsiveness strain (Roberts et al., 2000; Geiger et al., 2004). As the thalassemic mouse model we used in our experiments has been developed on a C57BL/6J genetic background (Yang et al., 1995), a direct comparison between C57BL/6 and HBBth-3 mice in terms of mobilization proficiency could be reliably undertaken, at the expense, however, of a lower response to G-CSF priming by either strain.
Our study has demonstrated that thalassemic mice mobilize less effectively than the normal strain. This could imply stem cell sequestration in the enlarged spleen, impaired release of HSCs from the BM to the bloodstream. or a reduced hematopoietic stem cell pool within the thalassemic BM. As there was no difference in the BM content of HSCs between normal and thalassemic mice both at steady state and after G-CSF priming, the possibility of stem and progenitor cell uptake by the spleen seemed prominent. On the other hand, the observed higher content of LSK cells and CFCs in the thalassemic spleen after G-CSF treatment could reflect increased in situ proliferation of stem cells, splenic trapping, or both. The thalassemic spleen after G-CSF treatment was shown by immunohistochemistry to be a site of both intense cell proliferation (Ki-67 positivity) and sequestration of stem and progenitor cells (c-Kit+ cells within sinusoids and trabecular vessels). Splenectomy restored the peripheralization of LSK cells and CFU-GM, suggesting that the thalassemic spleen primarily pools stem and progenitor cells from the periphery during G-CSF treatment rather than releases in situ-produced cells to the bloodstream.
At present, the only existing information abut mobilization efficacy in thalassemia is derived from the report of Li and colleagues (1999), in which most of the patients included were pediatric. This study showed comparable yield and composition of harvest between thalassemic children and normal donors with 1-day delay of thalassemic patients to achieve peak levels of CD34+ cells. This is in contrast to the significantly lower peripheralization of LSK cells after G-CSF treatment in our thalassemia intermedia mice as compared with the normal strain. This discrepancy between thalassemic mice and patients may be due to the disease phenotype (thalassemia major vs. intermedia), the splenectomy status of patients (not reported in the study by Li and colleagues), or the stem cell immunophenotypic definition used in each study (LSK cells vs. CD34+ cells), or it may reflect differences in mobilization between species (mice vs. humans).
G-CSF-related splenic enlargement (Platzbecker et al., 2001; Stroncek et al., 2003), which rarely may result in splenic rupture (Becker et al., 1997; Brown and Dale, 1997; Falzetti et al., 1999; O'Malley et al., 2003; Balaguer et al., 2004), has been documented in normal donors and patients with hematological malignancies. The exact mechanism of splenic enlargement and spontaneous rupture after G-CSF remains unclear. It is potentially caused by either extramedullary hematopoiesis and/or intrasplenic accumulation of HSCs, myeloid progenitors, and mature cells. G-CSF administration in a condition of preexisting splenomegaly, as in thalassemia, may theoretically carry an increased risk of splenic rupture. In our study, there was no clinical evidence of rupture of the spleen after G-CSF treatment in either strain. Surprisingly, however, we observed a significantly stronger effect of G-CSF in normal-sized spleens compared with thalassemic spleens, resulting in a relative increase in spleen size by a factor of 2.5 in the normal C57 strain and by a factor of 1.6 in the thalassemic model (133 vs. 65% increase, respectively). Picardi and colleagues (2003) have reported a significantly higher median increase in spleen volume in mobilized normal donors relative to patients with hematologic malignancies (122 vs. 66.5%, respectively). We and others (Gaia et al., 2006; Yannaki et al., 2006) have also reported a relatively low median increase in spleen size (10–20%; range, 0–78%) in cirrhotic patients after mobilization with G-CSF. It is possible that in conditions of chronic splenomegaly, in which splenic size fluctuates in response to the degree of extramedullary erythropoiesis or hypersplenism or leukemic infiltration, the spleen becomes more flexible compared with those of normal individuals. Thus, although G-CSF priming may cause a striking enlargement of a normal spleen over a short period of time, it may not result in abrupt changes in splenic size in an already enlarged spleen. In addition, we also observed a significant size increase in the thalassemic liver but not in the normal liver after G-CSF treatment, which was correlated with a G-CSF-induced increase in liver hematopoiesis of thalassemic mice, probably to compensate for the suppression of erythroid lineage in the bone marrow stimulated by G-CSF. In normal mice, G-CSF-induced extramedullary hematopoiesis was practically restricted to the spleen, with only minimal evidence of a switching of erythropoiesis to the liver. Limited compensatory liver erythropoiesis after G-CSF administration was found in the liver of normal mice only after splenectomy, as was also shown previously (Molineux et al., 1990). These data imply that a space saturation effect in the thalassemic spleen after G-CSF treatment might be involved: when saturation is reached, G-CSF-induced stimulation of hematopoiesis or/and cell migration is taken up by the liver. This hypothesis could help to interpret the observed stronger effect of G-CSF on the size of normal spleen relative to thalassemic spleen.
No splenic rupture was detected in either strain after G-CSF treatment and mobilization was overall well tolerated. However, intraparenchymal hemorrhagic infarcts were developed at low frequency (12.5%) in thalassemic spleens after G-CSF treatment, in contrast to the uneventful procedure reported by Li and colleagues (1999) in 20 mobilized patients with β-thalassemia major. The thalassemia intermedia phenotype of the (untransfused) mice in our study may have been the main contributor to the development of splenic infarcts during mobilization, by pooling from the circulation high numbers of damaged red cells. In addition, during G-CSF priming, the increased blood viscosity due to high numbers of activated circulating cells in the spleen, and the increased parenchymal congestion that impeded the blood flow within the spleen, may have further facilitated the formation of splenic infarcts. Microvascular flow disturbances have been described in a mouse model reproducing the major features of β-thalassemia (Stoyanova et al., 2007) and thromboembolic events have been documented in subjects with thalassemia, more prevalent in splenectomized subjects with thalassemia intermedia not receiving regular transfusions (Eldor et al., 1999; Eldor and Rachmilewitz, 2002). In addition, procoagulant effects precipitating thrombotic events in high-risk individuals have been associated with G-CSF (Lindemann and Rumberger, 1993; Fukumoto et al., 1997). According to our data and the safe mobilization procedure in pediatric patients (Li et al., 1999), the thrombotic complications during G-CSF mobilization in regularly transfused patients are predicted to be low, although not negligible.
The effect of G-CSF in erythropoiesis still remains a controversial issue. Long-term G-CSF treatment in mice results in depression of the erythroid lineage within the bone marrow without, however, causing anemia because of the compensatory increase in splenic erythropoiesis (de Haan et al., 1992). The lack of compensatory splenic erythropoiesis in splenectomized mice results in anemia (Cronkite et al., 1993). Conceivably, in humans in whom there is a lack of splenic erythropoiesis, G-CSF may result in or worsen anemia, by stimulating granulopoiesis at the expense of an erythroid lineage steal within the BM (Kojima et al., 1991; Papaldo et al., 2006). In contrast, there are reports of a positive effect of G-CSF on human erythropoiesis (Miles et al., 1990; Park et al., 1991). In our study, G-CSF administration did not affect the Hb level or reticulocyte counts in either strain. It is of interest, however, that although splenectomy reduced hemoglobin levels in the normal strain, both at steady state and after G-CSF treatment, it did not significantly affect Hb in the thalassemic strain, probably because of the development of liver compensatory hematopoiesis.
Given the restrictions derived from our preclinical thalassemia intermedia model, an extrapolation of our data to the human situation could be attempted, with the reservation that thalassemic patients with intact spleens may mobilize less efficiently than normal subjects. Pediatric patients with unknown spleen status were shown to mobilize HSCs comparably to normal donors in the report of Li and colleagues (1999); however, one may consider that adult thalassemia patients have more extensive organ damage than pediatric patients, due to chronic iron accumulation, and that the BM stem cell reservoir of these polytransfused and long-term chelated adults is unclear (Vlachaki et al., 2007). These parameters may adversely affect mobilization in terms of safety and efficacy. In a gene therapy setting, where high numbers of transduced HSCs need to be infused in order to effectively compete for niche in the thalassemic BM, either multiple mobilization cycles or alternative mobilization procedures, such as the combination of AMD3100 and G-CSF, may be required. The spleen status of the patients, by potentially affecting the final HSC yield, may also represent a variable that needs to be taken into consideration in designing a clinical protocol.
AMD3100 (plerixafor) reversibly disrupts the binding of stromal cell-derived factor (SDF)-1 to CXCR4, resulting in a decreased ability of stem cells to migrate toward and adhere to the bone marrow microenvironment, finally increasing the number of progenitors in the circulation (Liles et al., 2003; Devine et al., 2004). When added to G-CSF it has been shown to improve the mobilization yield by severalfold in normal donors and poorly mobilizing patients (Flomenberg et al., 2005; Liles et al., 2005; Calandra et al., 2008). Plerixafor may result in improved mobilization efficiency in patients with thalassemia by releasing stem cells to the bloodstream not only from the BM but also from extramedullary sites such as the spleen or the liver, thereby overcoming the limitation of splenic cell sequestration. To further support this hypothesis, plerixafor was shown to exert diminished mobilizing ability in asplenic mice in contrast to effective mobilization of HSCs in nonsplenectomized mice (Ramirez et al., 2009).
G-CSF-related splenic enlargement and/or the risk of rupture does not seem to be increased in this animal model of thalassemia. However, excessive splenic hematopoiesis along with activation of the cells by G-CSF may lead to splenic infarcts. Consequently, in view of human gene therapy of thalassemia, strategies to reduce extramedullary hematopoiesis as well as overall cell production (i.e., pretreatment with hydroxyurea, hypertransfusion) before G-CSF treatment should be explored in a clinical setting.
The safety and efficacy of G-CSF mobilization, with or without hydroxyurea pretreatment in adult splenectomized and nonsplenectomized patients with β-thalassemia major, is currently being explored in a clinical trial conducted at our institution (George Papanicolaou Hospital, Thessaloniki, Greece).
Footnotes
Acknowledgments
This work was supported by a grant from the 3rd European Community Framework, Operational Program for Competitiveness, Axis 4, Measure 4.5. Part of the study was presented at the 18th Greek Hematology Society Meeting, Thessaloniki, November 14–17, 2007, and received the Arkagathos Gouttas Award.
Author Disclosure Statement
The authors report no potential conflicts of interest to disclose.