
Abstract
Pancreatic cancer is the fourth leading cause of cancer-related death in the United States, and even under optimal therapy these patients face a poor prognosis. Here we report a novel gene therapy-based strategy to battle this disease. We show that the majority of pancreatic tumors overexpress c-erb-B2, which therefore might serve as a target for novel therapies. On the basis of these findings, we developed an adenoviral vector [Ad-e23(scFv)-PE40] encoding a c-erb-B2 (Her-2/neu)-targeted immunotoxin. To improve viral gene delivery we coinfected the therapeutic adenovirus with a replication-competent adenovirus (RCAd) at low doses that enhanced the transduction efficiency of the former virus. In addition, we show that target gene expression can be enhanced by adding etoposide (VP16) at nontherapeutic doses. To investigate the therapeutic efficacy of our approach we established a mouse model for advanced pancreatic cancer disease by intraperitoneal injection of pancreatic cancer cell lines, resulting in multifocal peritoneal xenograft tumors. Administration of Ad-e23(scFv)-PE40 in combination with RCAd and VP16 significantly inhibited tumor growth in mice, with no apparent systemic toxicity. In this study we show that c-erb-B2 might be an effective molecular target in the treatment of pancreatic tumors and that coadministration of a therapeutic c-erb-B2-targeted, non-replication-competent adenovirus with an RCAd and VP16 could be a powerful approach to effectively deliver therapeutic genes to tumors. As demonstrated, this strategy can be employed to effectively treat pancreatic cancer in particular, but may be modified to treat other types of cancer as well.
Introduction
Gene therapy is a promising strategy for cancer treatment in the clinic. Attempts have been made to enhance host immune responses, to restore tumor suppressor genes, and to inhibit oncogene expression by gene therapy, but its feasibility and efficacy in clinical cancer treatment have yet to be demonstrated (Cross and Burmester, 2006). One of the major obstacles is the low efficiency of gene delivery. A recombinant adenovirus (rAdV) is one of the most efficient vectors currently available for gene delivery. It can transduce a variety of normal and tumor cells because of the broad expression of the coxsackievirus and adenovirus receptor (CAR) required for rAdV infection. Both replication-deficient (RDAd) and replication-competent (RCAd) adenoviruses have been used for gene delivery; however, as replication-deficient adenoviruses are unable to replicate, reinfect, and spread, they are considered safer and, thus, are more commonly used.
Transport of RDAd through tumor tissues is limited because of the presence of physiological barriers. The transport issue is less of a concern for RCAd because it can spread to neighboring cells through killing of the host cells during its replication cycle. However, apart from safety issues there are some disadvantages with RCAd: The capacity of RCAd to carry therapeutic genes is limited because of their larger size. In addition, as host cells are eventually killed by viral lysis, the expression of therapeutic genes comes to an end whereas cells infected with a replication-incompetent virus continue to express the viral transgene for an extended period of time.
To take advantages of both vectors and minimize their disadvantages, researchers have developed a new strategy, in which a conditionally replicating adenovirus is used to induce replication of the RDAd by providing the E1 protein in infected tumor cells, which is required for viral replication (Lee et al., 2004, 2006). As a result, RDAd can spread throughout the tumor while still avoiding systemic dissemination.
Another approach to improve adenoviral gene delivery is to use chemotherapeutic agents to control or enhance the expression of therapeutic genes. Park and colleagues have reported that cisplatin could transcriptionally activate the early growth response-1 (Egr1) promoter, which could be used to control the expression of therapeutic genes (Park et al., 2002). Etoposide, also known as VP16, can enhance the efficiency of adenoviral infection through upregulating CAR expression (Shieh et al., 2006). In addition, VP16 is known to activate the promoters of a number of genes, such as p53, hypoxia-inducible factor-1α, and telomerase reverse transcriptase (Sato et al., 2000; Hussein et al., 2006). Therefore, if these promoters are used to control viral transgene expression, addition of VP16 will activate transcription of the transgene.
To reduce the adverse effects of rAdV in normal tissues, a tumor-specific targeted vehicle or therapeutic gene is desirable. Fortunately, tumor cells often overexpress unique receptors or antigens that distinguish them from surrounding normal tissues and can be excellent targets of novel vehicles or therapeutic genes. For example, members of the epidermal growth factor receptor (EGFR) family, most prominently EGFR/ErbB and its ligands (EGF, transforming growth factor TGF-α, and TGF-β), have been found to be elevated in many tumors, including pancreatic cancer (Yarden and Sliwkowski, 2001; Siegel and Massague, 2003; Cardenes et al., 2006; Hezel et al., 2006; Pino et al., 2006; Tzeng et al., 2007). EGFR inhibitors alone or in combination with cytotoxic chemotherapy can inhibit the growth and metastasis of human pancreatic cancer in vitro and in vivo (Bruns et al., 2000; Solorzano et al., 2001; Zalatnai, 2007; Azzariti et al., 2008), and have been approved for treatment of pancreatic cancer in patients (Czito et al., 2006). c-erb-B2 is another member of the EGFR family and is frequently upregulated in epithelium-derived tumors, such as breast, ovarian, and gastrointestinal carcinomas (Koeppen et al., 2001; Kim et al., 2007). Our data and other reports in the literature (Tsiambas et al., 2006) demonstrate that Her-2/neu is also overexpressed in pancreatic carcinoma, and that Her-2/neu protein expression is strongly associated with the grade of pancreatic tumor. These clinical data suggested that c-erb-B2 was an attractive molecular target in the development of a novel gene therapy strategy for the treatment of pancreatic carcinoma.
To selectively target c-erb-B2 and block its downstream activity, monoclonal antibodies have been developed and proven to be an effective adjuvant treatment for Her2/neu (c-erb-B2)-overexpressing breast cancer (Pritchard et al., 2006). The antitumor activity of anti-erb-B2 antibodies may be mediated by the ability of antibodies to shift the equilibrium from active dimeric to impaired tetrameric receptor complex states, leading to the formation of inactivated tetrameric forms of c-erb-B2 receptors (Yakes et al., 2002; Nagata et al., 2004; Furuuchi et al., 2007).
In addition to their cytotoxic effects, monoclonal antibodies can be used to deliver a recombinant immunotoxin specifically to cells expressing the respective antigen. The immunotoxin e23(scFv)-PE40 is a recombinant fusion protein consisting of a secretable single-chain antibody directed against c-erb-B2 and the 40-kDa intracellular domain of Pseudomonas exotoxin A (PE40), which was constructed to contain a signal peptide, a single-chain antibody derived from the monoclonal antibody e23, and the bacterial toxin PE40 lacking the binding domain. It takes advantage of the high selectivity and affinity of the antibody domain combined with the potent cytotoxicity of the bacterial toxin and can therefore be used to specifically kill cells expressing the targeted antigen. Because our data demonstrated that c-erb-B2 was overexpressed in a majority of human pancreatic carcinomas, this agent might be effective in pancreatic cancer treatment.
Materials and Methods
Human normal tissue array and pancreatic specimens
Human normal tissue arrays were purchased from the National Engineering Center for Biochips at Shanghai (Shanghai Outdo Biotech, Shanghai, China), and contained 20 different types of normal human tissue, including cerebrum, cerebellum, brainstem, tongue, esophagus, stomach, intestine, appendix, liver, pancreas, thyroid, heart, aorta, lung, trachea, bladder, prostate, testis, skin, and muscle. Ten normal pancreatic tissues were obtained from the First People's Hospital of Shanghai Jiaotong University (Shanghai, China).
Patients and pancreatic tumor specimens
Thirty-six tumor specimens were obtained from patients pathologically diagnosed with pancreatic adenocarcinoma and enrolled in this study (26 of the specimens were derived from patients at the First People's Hospital of Shanghai Jiaotong University and 10 from patients at General Hospital, West China University of Medical Sciences). The clinical parameters of the 36 patients with pancreatic adenocarcinoma are summarized in Supplementary Table 1 (see
*The scoring was done in five high-power views for each slide and 100 cells per view were counted by two experienced pathologists. The scoring was as follows: —, no appreciable staining in normal cells; +, barely detectable staining in cytoplasm and/or nucleus compared with stromal elements; ++, readily appreciable brown straining distinctly marking tumor or normal cells cytoplasm and/or nucleus; +++, dark brown staining in normal cells completely obscuring cytoplasm and/or nucleus.
Cell lines
Three human pancreatic tumor cell lines, SW1990, PANC-1, and Bxpc-3, were used in this study. They were obtained from the cell bank at the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). An adenovirus-packaging cell line (AD-293) was purchased from Stratagene (La Jolla, CA). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) or RPMI 1640 containing 10% fetal bovine serum (FBS),
Immunohistochemical staining
The primary antibodies for human c-erb-B2 and CAR were purchased from Lab Vision/Thermo Fisher Scientific (Fremont, CA; Neomarkers, clone e2-4001 + 3B5, diluted 1:400) and Upstate Cell Signaling Solutions/Millipore (Lake Placid, NY; clone RmcB, diluted 1:10), respectively. Immunohistochemical staining was carried out according to the manufacturers' protocols. Expression levels of c-erb-B2 and CAR were visualized with 3,3′-diaminobenzidine (Dako, Glostrup, Denmark). Scoring was carried out in five fields of view, each containing at least 100 cells, by two experienced pathologists referring to published reports (Koeppen et al., 2001; Korn et al., 2006; Kim et al., 2007).
Western blotting
Expression of c-erb-B2 in pancreatic tumor cell lines was determined by Western blotting. The cells were lysed, and total protein concentration was determined with the Bradford reagent. About 60 μg of each sample was subjected to electrophoresis in sodium dodecyl sulfate (SDS)–10% polyacrylamide gels and transferred to a polyvinylidene difluoride transfer membrane. The membranes were probed with primary monoclonal antibodies against c-erb-B2 (Neomarkers, diluted 1:1000) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (diluted 1:5000; Kangchen, Shanghai, China) as loading control as well as appropriate secondary antibodies conjugated with horseradish peroxidase. The immunoreactive bands were visualized with Lumi-Light Western blotting substrate (Roche, Mannheim, Germany).
Adenoviruses
Ad-e23(scFv)-PE40 for production of c-erb-B2 (Her-2/neu)-targeted secretable immunotoxin was constructed by homologous recombination as described by Microbix (Toronto, ON, Canada). Briefly, DNA fragment e23(scFv)-PE40, which is driven by the cytomegalovirus (CMV) promoter and encodes a fusion protein consisting of a secretable single-chain antibody against c-erb-B2 and a 40-kDa intracellular domain of Pseudomonas exotoxin A (PE40), was inserted into the shuttle plasmid pDC316 and cotransfected into AD-293 cells along with plasmid pBHGloxΔE1.3cre carrying the adenoviral genome. After homologous recombination between pDC316-e23(scFv)-PE40 and pBHGloxΔE1.3cre, an E1-deleted, replication-deficient type 5 adenovirus was produced. Well-isolated plaques were selected through titrated infectivity on monolayer AD-293 cells and further screened by polymerase chain reaction (PCR) and Western blotting for the gene of interest. Several other adenoviruses were developed in our laboratory. They include Ad-EGFP (an E1-deleted, replication-deficient adenovirus with CMV promoter-driven enhanced green fluorescent protein [EGFP] expression cassette), Ad-B7-mIL12 (an E1-deleted, replication-deficient adenovirus with CMV promoter-driven B7 and mIL-12 expression cassette), and Ad-TERT-E1A/E1B (a replication-competent adenovirus with telomerase reverse transcriptase promoter-driven E1A/E1B expression cassette and CMV promoter-driven Discosoma red fluorescent protein [dsRFP] expression cassette, the RCAd used in this paper) (Huang et al., 2004). The titer of each adenovirus was determined in terms of plaque-forming units (PFU) in AD-293 cells.
Transduction efficiency of adenoviral vector in vitro
SW1990 tumor cells were infected for 72 hr with Ad-EGFP with or without RCAd or VP16. Expression of EGFP and RFP was assessed with an inverted epifluorescence microscope (Carl Zeiss, Göttingen, Germany) and recorded, and the status of infected cells was assessed by bright-field illumination and recorded. The EGFP positivity rate and average fluorescence intensity (MnX) were further assessed by flow cytometric analysis (Beckman Coulter, Fullerton, CA), and living cells were assessed with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium/phenazine methosulfate (MTS/PMS) assay kits in accordance with the instructions of the manufacturer (Promega, Madison, WI). All assays were performed in three individual wells and repeated three times. The mean from triplicate assays is reported in Results.
Cytotoxicity assay in vitro
To determine the cytotoxicity of the immunotoxin molecule e23(scFv)-PE40 in vitro, we directly applied various amounts of Ad-e23(scFv)-PE40 or Ad-EGFP with or without RCAd or VP16 to tumor cells. Viable cells were counted 24, 48, 72, and 96 hr postinfection via MTS/PMS assay, or the density of living cells was visualized by violet staining.
Apoptosis assay in vitro and in vivo
Apoptotic cells were identified by annexin V-binding assay or terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) assay. For the annexin V-binding assay, human pancreatic tumor SW1990 or PANC-1 cells were plated in 6-well plates and infected with Ad-e23(scFv)-PE40 or Ad-null at multiplicities of infection (MOIs) of 3 and 6, respectively, for 48 hr. Annexin V-positive cells were identified by flow cytometry (FCM) according to the manufacturer's instructions. For the TUNEL assay, SW1990 or PANC-1 cells were plated on coverslips in 24-well plates and infected with Ad-e23(scFv)-PE40 or Ad-null at MOIs of 3 and 6, respectively, for 48 hr. After being fixed with 4% paraformaldehyde, apoptotic cells on slides were subjected to TUNEL assay as described in the manufacturer's protocol (Takara, Dalian, China) and visualized with 3,3′-diaminobenzidine. For the detection of apoptotic cells in xenografted tumors, formalin-fixed tumor sections were deparaffinized, rehydrated, and subjected to TUNEL staining as described previously.
Quantitative real-time PCR
About 7 × 105 SW1990 tumor cells were plated into 6-well plates, and infected with either 3 MOI of Ad-e23(scFv)-PE40 alone or with the same amount of adenoviral vector plus VP16 (20 μg/ml) or 0.6 MOI of RCAd for 48 or 72 hr. Total RNA was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA concentration was determined by spectrophotometry. Reverse transcription was carried out with random hexamers, using a SuperScript FIRST-strand synthesis kit (Invitrogen), and then the secondary strand was synthesized according to the manufacturer's instructions.
The following primers were used: For e23(scFv)-PE40, 5′-CAAGGGCAAGGCCACATT-3′ and 5′-GGCGATGACGGGTGAAAGT-3′; for GAPDH, which was used as an internal control, 5′-GAAGGTGAAGGTCGGAGT-3′ and 5′-GAAGATGGTGATGGGATTTC-3′.
For quantitative real-time PCR, the SYBR green system was used. A total PCR volume of 20 μl was prepared with distilled H2O, containing 0.1 μl of cDNA from the reverse transcription (RT), a 0.5 μM concentration of each primer, and 10 μl of reagent from the Platinum SYBR green qPCR supermix UDG kit (Invitrogen). Quantitative PCRs were performed with a DNA Engine Opticon 2 (MJ Research/Bio-Rad, Waltham, MA) with an initial denaturation step at 95°C for 2 min, followed by 40 cycles of denaturing at 94°C (10 sec), annealing at 55°C (30 sec), and extension at 72°C (30 sec), and completed by polymerization at 72°C (5 min).
Genomic DNA was extracted from the infected SW1990 tumor cells to determine the copy number of e23(scFv)-PE40, using a universal genomic DNA extraction kit (Takara), and then subjected to quantitative PCR as described previously. GAPDH was amplified as internal control for DNA input. Data were analyzed using the comparative ∆∆ cycle threshold (2–∆∆CT) model, where ∆∆C T is defined as [C T, e23(scFv)-PE40 − C T, GAPDH]sample/[C T, e23(scFv)-PE40 − C T, GAPDH]control (Livak and Schmittgen, 2001; Fleige et al., 2006). mRNA or genomic DNA in cells infected with Ad-e23(scFv)-PE40 alone was used as control.
Expression of e23(scFv)-PE40 in SW1990 cells infected with 3 MOI of Ad-e23(scFv)-PE40 alone or the same amount of adenoviral vector plus VP16 (20 μg/ml) or 0.6 MOI of RCAd was determined by Western blotting with anti-Pseudomonas exotoxin A (anti-PEA) antibody (diluted 1:200; Sigma-Aldrich, St. Louis, MO).
Evaluation of serum concentration of murine interleukin-12
All experimental animals were obtained from the Animal Research Committee of the Institute of Biochemistry and Cell Biology (Shanghai, China). Approximately 5 × 108 PFU of Ad-B7-mIL12 plus 1 × 108 PFU of RCAd or 5 × 108 PFU of Ad-B7-mIL12 alone in a total of 100 μl of sterile phosphate-buffered saline (PBS) was injected into either the tail vein, peritoneal cavity, or hind leg muscle of 6-week-old male C57BL/6 mice (n = 4). On days 1, 3, 6, and 8 after adenovirus injection, blood was collected from the caudal vein, and the serum concentration of murine interleukin (mIL)-12 was quantified with an mIL-12 ELISA kit (R&D Systems, Minneapolis, MN).
Efficacy of tumor gene therapy in vivo
SW1990 cells (2 × 106) suspended in 200 μl of PBS were injected into the peritoneal cavity of 6-week-old male athymic BALB/c nude mice to establish a human pancreatic xenograft model. The model mimicked intraperitoneal metastases of late-stage pancreatic carcinoma. On the fourth day after tumor cell implantation, mice with the human pancreatic carcinoma xenograft were divided into seven groups (n = 6): Ad-e23(scFv)-PE40 alone, RCAd alone, VP16 alone, Ad-e23(scFv)-PE40 plus RCAd, Ad-e23(scFv)-PE40 plus VP16, Ad-e23(scFv)-PE40 plus RCAd and VP16, and PBS. The doses of adenovirus and VP16 were as follows: 5 × 108 PFU of Ad-e23(scFv)-PE40, 1 × 108 PFU of RCAd, and 800 μg of VP16 in 200 μl of PBS. They were administered into the peritoneal cavity of each mouse on days 4, 6, and 8 after tumor cell implantation. The weight of mice was measured periodically. On day 14, the mice were killed and the number and weight of the disseminated tumors in the peritoneal cavity were measured and recorded. Tumor specimens were fixed in formalin and embedded in paraffin. Deparaffinized tumor sections were subjected to TUNEL assay and immunohistochemical assay as described previously. Adjacent sections were stained with hematoxylin and eosin (H&E).
Examination of serum biochemical markers
When mice were killed on day 14, blood was collected for biochemical analysis. Blood parameters analyzed included alanine aminotransferase (ALT), alkaline phosphatase (ALP), aspartate aminotransferase (AST), urea, total serum bilirubin (TBIL), and creatine kinase (CK). The examination was performed with Roche Modular Analytics SWA P800 (Roche Diagnostics, Indianapolis, IN).
Statistical analysis
All statistical analyses were conducted with SPSS 11.5 software (SPSS, Chicago, IL). One-way analysis of variance (ANOVA) was performed for analysis of cytotoxicity, tumor weight, and serum biochemical markers; Fisher's exact test was used for analysis of clinical data. p < 0.05 was considered significant.
Results
Differential c-erb-B2 and CAR expression in normal human tissues
Targeted therapies rely on differential expression of their target (i.e., the antigen to which they are targeted) in normal and tumor tissues. When designing a targeted therapy it is therefore important to map expression profiles of the respective antigen in normal tissue as well as in tumor tissue. We subjected 20 different normal human tissues to immunohistochemical analysis of c-erb-B2 expression; the results are shown in Table 1. Representative images of immunohistochemical stainings in normal tissues are shown in Fig. 1A–D. Of all the tissues analyzed, 12 samples were found to stain positively for c-erb-B2, albeit with variable intensity: neurons in cerebrum; Purkinje's cells in cerebellum; cells in brainstem; myocytes in tongue, heart, and skeletal muscle; epithelium in esophagus, stomach, intestine, appendix, and bladder; basal cells and sebaceous gland in skin; and pancreatic islets. Acinar and ductal cells of normal pancreas did not exhibit any c-erb-B2 staining. Liver, lung, trachea, prostate, testis, thyroid, and aorta were also found to be negative for c-erb-B2 expression.

c-erb-B2 and coxsackievirus and adenovirus receptor (CAR) expression in normal human tissues, pancreatic tumor tissues, and cell lines. Immunohistochemical staining was carried out with monoclonal antibodies directed against c-erb-B2 and CAR, respectively. (
As adenoviruses show a preference for CAR-expressing cells, we determined the expression status of CAR in normal tissue. CAR was expressed in all normal human tissues tested except cerebrum, cerebellum, and aorta. CAR expression was high in esophagus and gastric epithelium, prostate, and myocytes of tongue; intermediate in brainstem, tracheal epithelium, intestinal epithelium, pancreatic islets, testis, and myocytes of the heart and skeletal muscle; and low in liver, lung, thyroid, bladder, skin, glandular epithelium of the appendix, and exocrine acinar and ductal cells of the pancreas (Table 1 and Fig. 1E–H). The c-erb-B2 and CAR expression pattern of the pancreas was further confirmed in another 10 normal pancreatic tissues.
c-erb-B2 and CAR expression in pancreatic carcinoma tumor specimens and cell lines
We analyzed 36 specimens of pancreatic adenocarcinoma for c-erb-B2 and CAR expression (shown in Fig. 1). Twenty-seven of 36 (i.e., 75%) pancreatic adenocarcinoma specimens were found to be c-erb-B2 positive; however, the percentage of positive cells in each sample and the intensity of staining varied significantly among the 27 samples. Representative images with weak and strong staining of c-erb-B2 are shown in Fig. 1I and J, respectively; the number of samples in each of these categories is given in Fig. 1M. In contrast to acinar and ductal cells in normal pancreatic tissue, adenocarcinoma cells of pancreatic tumors exhibited positive staining for c-erb-B2 (see Fig. 1I and J). In addition, c-erb-B2 expression in three pancreatic carcinoma cell lines was determined by Western blot analysis. c-erb-B2 expression was detected in all cell lines; however, expression levels varied significantly, as shown in Fig. 1O. CAR expression was observed in 20 of 36 (55.6%) pancreatic adenocarcinoma tumor tissues, again displaying some heterogeneity among the different samples. Representative images with weak and intermediate staining of CAR are shown in Fig. 1K and L, respectively; and the number of samples in each of these categories is given in Fig. 1N.
These data suggest that c-erb-B2 might be an attractive molecular target for gene therapy of pancreatic carcinomas and that, because of their positive CAR expression, pancreatic adenocarcinomas might be receptive to therapeutic adenoviruses.
Selective killing of pancreatic tumor cells by Ad-e23(scFv)-PE40 is c-erb-B2 related
To target pancreatic c-erb-B2-expressing tumor cells, we have engineered a replication-deficient adenovirus carrying the coding sequence of a secretable single-chain antibody against c-erb-B2 fused to the intracellular domain of Pseudomonas exotoxin A.
We investigated the cytotoxicity of Ad-e23(scFv)-PE40 alone at various doses (1, 3, 6, and 12 MOI) by MTS/PMS assay. As a control we infected cells with Ad-EGFP, a replication-incompetent adenovirus encoding EGFP, thus allowing us to assess transduction efficiency by reading out the GFP signal. The relative viability of SW1990 cells treated with Ad-e23(scFv)-PE40 was significantly lower compared with PANC-1 cells treated at doses of 3, 6, and 12 MOI (p < 0.01) (Fig. 2A).
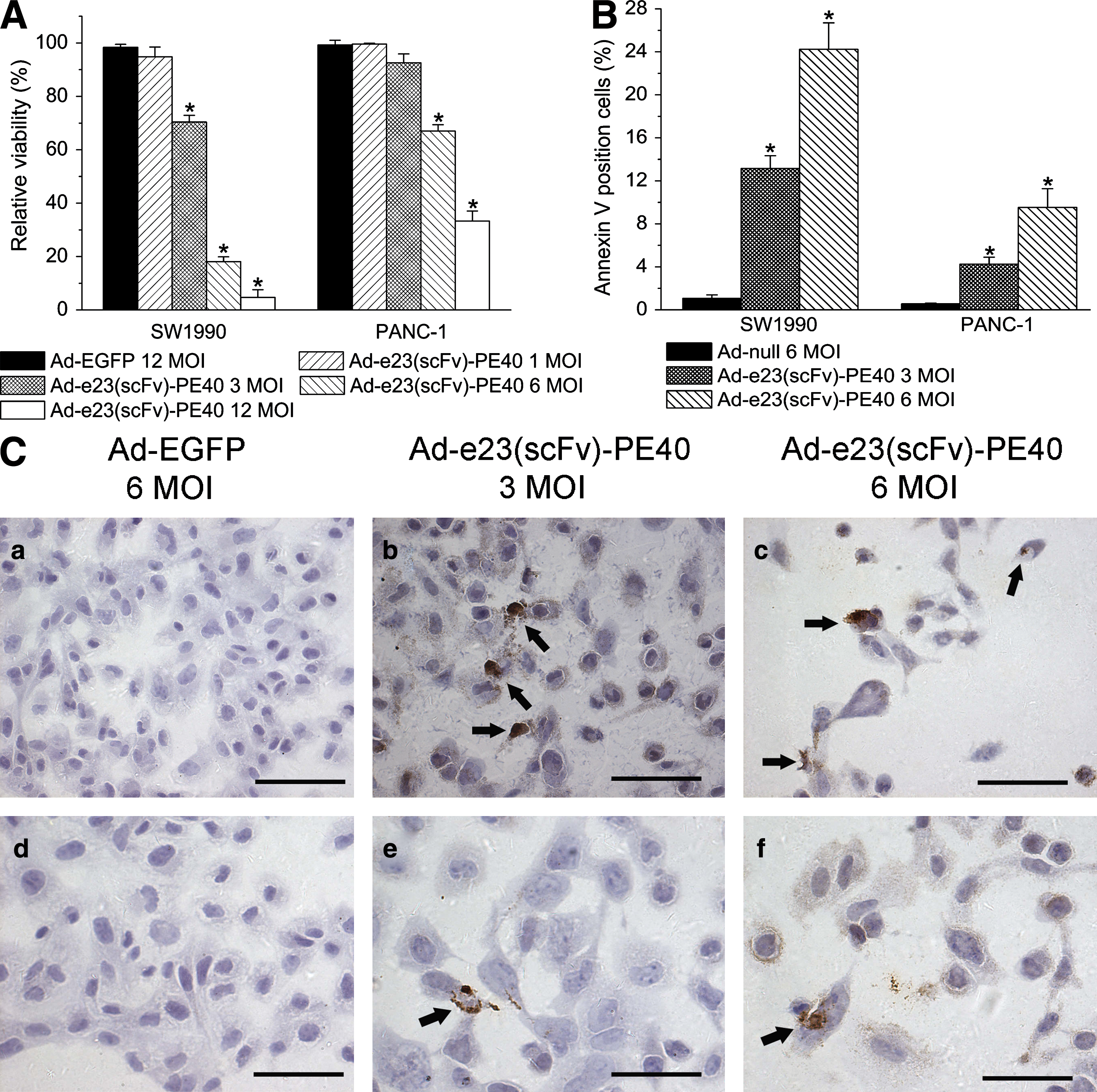
Antitumor effects of Ad-e23(scFv)-PE40 in vitro. (
To better characterize the cytotoxic effect of e23(scFv)-PE40, we quantified the rate of apoptosis on the basis of annexin V staining in cells transduced with either the therapeutic adenoviral vector Ad-e23(scFv)-PE40 or a control vector, Ad-null. Quantitative analysis was carried out by flow cytometry. Ad-e23(scFv)-PE40 induced apoptosis in a dose-dependent manner. In addition, the level of c-erb-B2 expression correlated well with the apoptosis rate in Ad-e23(scFv)-PE40-infected cells but not in cells infected with Ad-null (Fig. 2B). These results were confirmed by TUNEL staining, which can be used to detect later stages of apoptosis. As shown in Fig. 2C, SW1990 cells treated with Ad-e23(scFv)-PE40 showed significant apoptosis (brown staining) and detached from the slide at high doses (Fig. 2C, panels b and c), whereas SW1990 cells infected with 6 MOI of Ad-EGFP showed no significant apoptosis or morphologic changes (Fig. 2C, panel a). PANC-1 cells were less responsive to Ad-e23(scFv)-PE40 treatment than SW1990, presumably because c-erb-B2 expression was high on SW1990 cells and low on PANC-1 cells. These data demonstrate that Ad-e23(scFv)-PE40 induces apoptosis selectively in cells expressing c-erb-B2 and that the cell-killing effect was dependent on both the dose of Ad-e23(scFv)-PE40 virus and the expression level of c-erb-B2.
RDAd transduction efficiency can be enhanced by coinfection with replication-competent adenovirus
As transduction efficiency is a critical determinant of the therapeutic efficacy of a targeted adenovirus, we aimed to enhance transduction efficiency by coinfection with a replication-competent adenovirus that can provide the E1 gene required for viral replication. In addition, we used VP16 at low doses to enhance transgene expression either by regulating the promoter or by enhancing viral transduction efficiency by upregulating CAR expression.
To test whether these strategies are effective in enhancing transduction efficiency, we either coinfected a replication-deficient adenovirus for EGFP (Ad-EGFP) with a replication-competent adenovirus delivering red fluorescent protein (RFP) (RCAd-RFP) or added VP16 to the tumor treatment. Transgene expression and thus transduction efficiency was quantified by reading out the average GFP signal intensity as well as the percentage of GFP-positive cells, using flow cytometry. The results are shown in Fig. 3A, panels a and b, demonstrating that the amount of transgene expression, in an Ad-EGFP dose-dependent manner, was related to the dose ratios of Ad-EGFP and RCAd and of Ad-EGFP and VP16. For a given dose of Ad-EGFP, the optimal dose for enhancing EGFP gene delivery was between 0.3 and 1.2 MOI for RCAd and >20 μg/ml for VP16. When the dose of RCAd-RFP was increased to 4.8 MOI, EGFP expression was significantly reduced. The reduction was likely to be caused by RCAd-induced cell death because the viability of cells decreased with increasing doses of RCAd (see Fig. 3Ac).
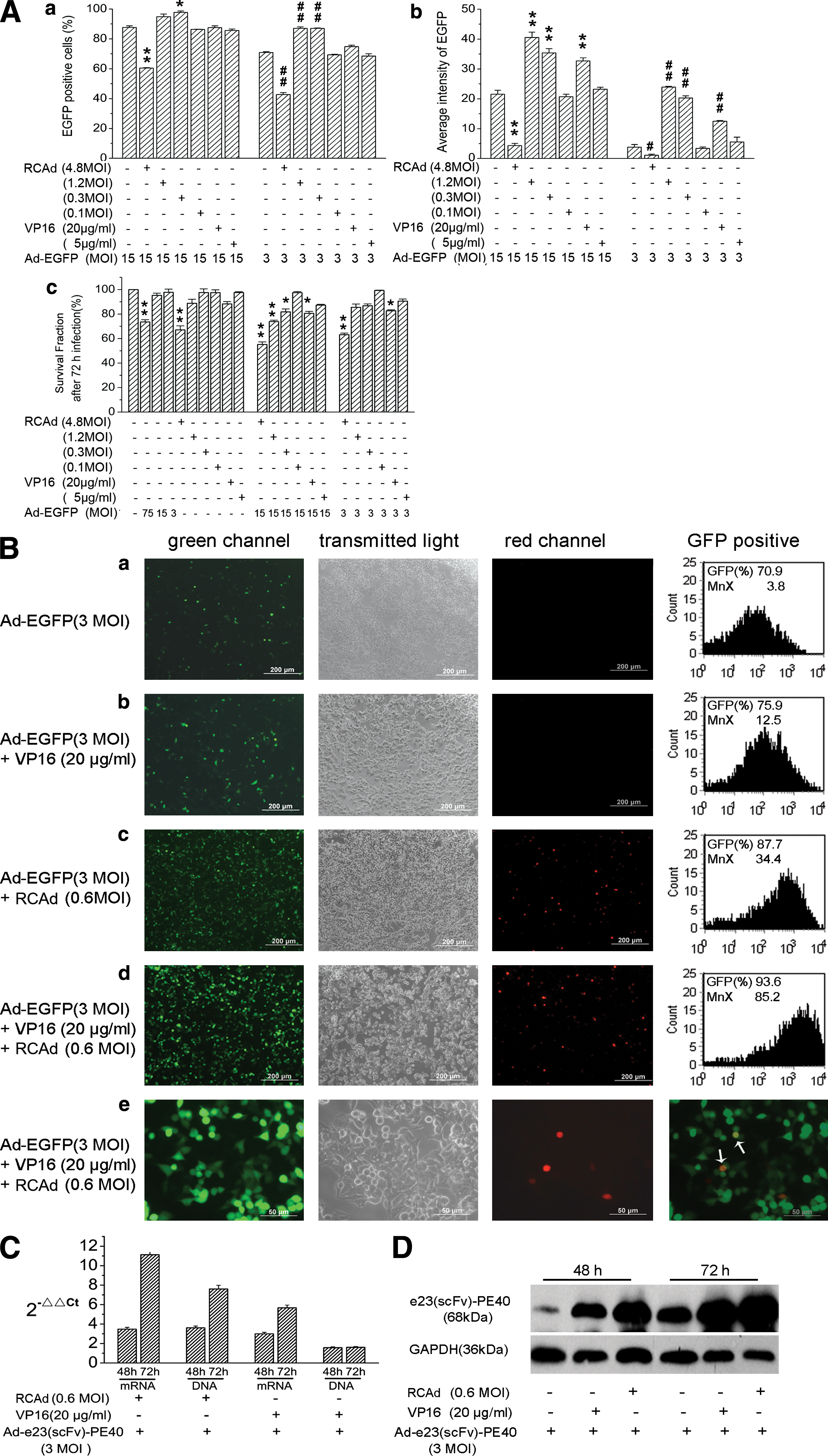
Replication-competent adenovirus (RCAd) or etoposide (VP16) enhanced replication-deficient adenovirus (RDAd)-mediated gene delivery to human pancreatic tumor cells. (
Representative images of cells infected with Ad-EGFP alone, Ad-EGFP plus VP16, Ad-EGFP plus RCAd-RFP, and Ad-EGFP plus VP16 and RCAd-RFP are shown in Fig. 3B, panels a–d, respectively. The flow cytometry data are shown in the corresponding panels for quantitative comparisons. These results further demonstrate that RCAd, VP16, or their combination can increase both the percentage of EGFP-positive cells and the average green fluorescence intensity of all cells and thus transduction efficiency. Interestingly, coinfection of RCAd-RFP could significantly increase the total number of EGFP-positive cells, although only a small fraction of cells actually coexpressed EGFP and RFP at the same time (Fig. 3B, panel e). The mechanism concerning how RCAd can affect RDAd-mediated gene delivery without coinfecting the same cell is currently unclear. The E1A protein in RCAd-infected cells might spread to adjacent cells to support RDAd replication. However, more studies need to be performed to support this hypothesis.
We then moved on to confirm that addition of RCAd and/or VP16 could indeed enhance gene delivery of RDAd e23(scFv)-PE40 by determining its DNA, mRNA, and protein levels in infected tumor cells. SW1990 cells were treated with either Ad-e23(scFv)-PE40 alone at 3 MOI, Ad-e23(scFv)-PE40 at 3 MOI plus VP16 at 20 μg/ml, or Ad-e23(scFv)-PE40 at 3 MOI plus RCAd at 0.6 MOI for 48 or 72 hr. After treatment, the tumor cells were analyzed by quantitative PCR (qPCR), RT-PCR, and Western blotting. The results shown in Fig. 3C and D indicated that both RCAd and VP16 could enhance Ad-e23(scFv)-PE40 gene delivery to a similar extent as shown previously for Ad-EGFP (see Fig. 3A and B).
As expected, addition of RCAd enhanced both Ad-e23(scFv)-PE40 replication and transgene expression whereas VP16 could only enhance transgene expression (Fig. 3C and D).
RCAd and VP16 enhance the cell-killing effect of Ad-e23(scFv)-PE40 in vitro
The results shown in Fig. 3 demonstrated that both VP16 and RCAd could significantly improve the transduction efficiency of RDAd. The next question concerned whether these agents could enhance the cell-killing ability of RDAd. We used Ad-e23(scFv)-PE40 (RDAd) to treat pancreatic tumor cell lines with various levels of c-erb-B2 expression: SW1990 (high expression) and PANC-1 (lower expression). When Ad-e23(scFv)-PE40 was combined with RCAd, VP16, or both, its therapeutic efficacy was enhanced in all cases (see Fig. 4). The level of enhancement was highly dependent on the doses of these agents and on the cell line used. For SW1990 cells, significant cell killing occurred in 6 of 24 experimental groups, in which the tumor cells were treated with Ad-e23(scFv)-PE40 alone at a high dose (6 MOI), Ad-e23(scFv)-PE40 at all doses tested plus RCAd (≥0.3 MOI), Ad-e23(scFv)-PE40 at all doses tested plus VP16 (≥20 μg/ml), or Ad-e23(scFv)-PE40 at all doses tested plus RCAd (≥0.3 MOI) and VP16 (≥5 μg/ml).

RCAd and VP16 enhanced the antitumor effects of Ad-e23(scFv)-PE40 in vitro. The cytotoxic effects of Ad-e23(scFv)-PE40 were augmented by both RCAd and VP16. SW1990 and PANC-1 cells were treated with either Ad-e23(scFv)-PE40, RCAd, Ad-EGFP, or VP16 alone or with a combination of these agents. Cells were cultured in 24-well plates and treated as indicated in Table 1. At 72 hr posttreatment, cells were stained with crystal violet, displaying the amount of viable cells by the extent of purple staining. Color images available online at
The efficacy of the gene therapy was higher for SW1990 cells than for PANC-1 cells, which is in concordance to the results shown in Fig. 2, where tumor cells were treated with Ad-e23(scFv)-PE40 alone. These data again indicate that the efficacy of the combined gene therapy depended on the level of c-erb-B2 expression on tumor cells.
The mechanisms of enhanced cell killing in combined gene therapy were unlikely to be due to direct cytotoxicity of RCAd and VP16 because if used individually without Ad-e23(scFv)-PE40, they had only minor effects on tumor cell density. The data shown in Fig. 3C and D suggested that the enhanced cell killing was caused by RCAd-mediated replication of Ad-e23(scFv)-PE40 and increased e23(scFv)-PE40 expression or by a VP16-mediated increase in e23(scFv)-PE40 expression.
Impact of administration route on serum concentration of transgene product
To determine the best administration route of our gene therapy we quantified the serum concentration of mIL-12 after administration of an adenovirus encoding mIL-12 (Ad-B7-mIL12) by either tail vein injection (intravenous), intraperitoneal injection, or hind leg muscle injection (intramuscular).
We administered 5 × 108 PFU of Ad-B7-mIL12 plus 1 × 108 PFU of RCAd or 5 × 108 PFU of Ad-B7-mIL12 alone in 100 μl of PBS to C57BL/6 mice (n = 4) and blood samples were collected 1, 3, 6 and 8 days after injection for determination of serum concentrations of mIL-12. The results are shown in Fig. 5A. The data demonstrated that the serum concentration strongly depended on the administration route. Its peak level was low after intramuscular injection, intermediate after intravenous injection, and high after intraperitoneal injection. When Ad-B7-mIL12 and RCAd were coadministered into the animals, the peak concentration after intraperitoneal injection was increased 2.5-fold as compared with intravenous injection. Interestingly, RCAd-augmented transgene expression was more significant when both adenoviral vectors were administrated intraperitoneally. It was minimal after intravenous injection. After intraperitoneal injection the serum concentration decreased more rapidly than in the other groups. As a result, on day 3 postinjection the serum concentration of mIL-12 was lower in the intraperitoneal injection groups than in the intravenous and intramuscular injection groups.

Transgene serum concentration, systemic toxicity, and RCAd- or VP16-enhanced antitumor efficacy of Ad-e23(scFv)-PE40 in vivo. (
RCAd- and VP16-mediated enhancement of Ad-e23(scFv)-PE40 gene therapy in vivo
To investigate the antitumor efficacy of Ad-e23(scFv)-PE40 in vivo, we established a human pancreatic xenograft model in nude mice by intraperitoneal implantation of 2 × 106 SW1990 cells. The model mimicked intraperitoneal metastases of late-stage pancreatic carcinoma. During the first few days a single, solid tumor mass was observed, which adhered to the lesser omentum of the pancreas. Micrometastases in the mesentery became visible 10 days after cell implantation. Gene therapy was started on day 4 after cell implantation. It was performed by intraperitoneal injection of approximately 5 × 108 PFU of Ad-e23(scFv)-PE40, 1 × 108 PFU of RCAd, and 800 μg of VP16 either alone or in various combinations. All group treatments were repeated on days 6 and 8, respectively. As it is difficult to monitor tumor growth in vivo we recorded the body weight of mice every other day, which has been shown to be an effective method to estimate tumor burden (Johnen et al., 2007). It was observed that body weight loss was fastest in the PBS-treated mice (Fig. 5B). On day 14 after cell implantation the mice were killed and the tumor masses in the peritoneal cavity were collected, measured and weighed. The results shown in Fig. 5C demonstrate that Ad-e23(scFv)-PE40 alone could significantly reduce the tumor mass in mice compared with PBS or treatment with RCAd and VP16 only. Tumor weight was significantly lower in groups where RCAd (p = 0.045), VP16 (p < 0.001), or both RCAd and VP16 (p < 0.001) was added to treatment with Ad-e23(scFv)-PE40 compared with Ad-e23(scFv)-PE40 treatment alone.
We also investigated the toxicity induced by intraperitoneal injection of Ad-e23(scFv)-PE40, with or without RCAd and/or VP16. In this study, blood samples were collected from the orbital sinus before mice were killed, and common biochemical markers in these samples were examined. The results are shown in Fig. 5D. There was no significant difference (p > 0.05) for all tested biochemical markers between the control group and the treated groups, indicating that intraperitoneal injection of adenoviral vector caused no obvious pathologic changes in mice.
As our gene therapy was targeted primarily at c-erb-B2-expressing cells, we examined c-erb-B2 expression in tumor sections after treatment. As expected, tumor sections of mice treated with Ad-e23(scFv)-PE40 or Ad-e23(scFv)-PE40 plus RCAd and/or VP16 showed significantly less expression of c-erb-B2 compared with the PBS-treated control group, indicating that our therapy selectively killed cerb-B2-expressing cells, resulting in a predominance of c-erb-B2-negative cells. In addition, TUNEL staining was performed to quantify the apoptotic rate in treated tumor sections. All analyzed tumor sections exhibited TUNEL staining; however, the staining was more pronounced in tumors that had been treated with Ad-e23(scFv)-PE40 or Ad-e23(scFv)-PE40 plus RCAd and/or VP16, reflecting the efficacy of our gene therapy. In addition, treatment with Ad-e23(scFv)-PE40 plus RCAd and/or VP16 resulted in more intensive necrosis throughout tumor sections (Fig. 6).

Detection of apoptosis and c-erb-B2 expression in xenograft tumor tissues after treatment. Shown are representative tumor sections of xenograft tumor treated with (
Discussion
The goal of this study was to develop a novel gene therapy approach to the treatment of pancreatic cancer. In this proof-of-concept study, we used this approach to treat an intraperitoneal pancreatic tumor model. The study was accomplished in three steps. The first was to identify unique molecular targets in pancreatic tumors for reducing normal tissue toxicity in gene therapy; to this end we systematically screened CAR and c-erb-B2 (a member of the EGFR family) expression levels in situ in 20 different types of normal human organs or tissues and 36 pancreatic tumor specimens. Our data suggested that c-erb-B2 could be a unique molecular target in pancreatic tumors. The second step was to develop a novel adenoviral vector to produce c-erb-B2 (Her-2/neu)-targeted immunotoxin locally in pancreatic tumors. The third was to investigate how to combine a low dose of RCAd or VP16 with RDAd for improving gene delivery into tumor cells. We also investigated effects of administration routes (intraperitoneal, intramuscular, and intravenous) on gene delivery to tumors as well as systemic toxicity in terms of the immunotoxin concentration in serum. The tumor models used in this study included cultured tumor cells and a human pancreatic xenograft implanted intraperitoneally in nude mice, which mimicked intraperitoneal disseminations of late-stage pancreatic carcinoma. Our data showed that the new gene therapy strategy could be used to improve treatment of pancreatic cancer as well as other abdominal neoplastic diseases.
Recombinant adenovirus (rAdV) is the most commonly used and most efficient gene delivery vehicle. Its transduction efficiency depends on binding to receptors (CAR, integrin, CD46) on the surface of cells or to coagulation factor X (FX) of cells (Waddington et al., 2008). CAR was initially identified for cell infection by group B coxsackieviruses (Honda et al., 2000; Cohen et al., 2001). It was later identified to be a primary receptor of certain types of human adenovirus (Bergelson et al., 1997; Coyne and Bergelson, 2005), including the most popular gene delivery vehicles, Ad5 and Ad2. Therefore, CAR expression is an important determinant of a cell's susceptibility to infection and transduction by Ad5 and Ad2. Previous RNA blot analysis had suggested that in adults, CAR is significantly expressed in heart, testis, prostate, brain, pancreas, and intestine (Tomko et al., 1997; Bergelson et al., 1998) as well as in lung and liver (Fechner et al., 1999). An immunohistochemical staining analysis suggested that CAR protein can be detected in normal gastrointestinal tissues, such as colonic and esophageal mucosae, pancreas, and liver (Korn et al., 2006). However, the systematic screening of CAR protein expression in situ in human tissues and tumor specimens by immunohistochemistry has been rarely reported, although it is important for improving adenoviral vector-mediated gene therapy. Therefore, we examined CAR expression in 20 different types of human tissue by immunohistochemical staining. We found that CAR was expressed at various levels in almost all human tissues tested. Interestingly, CAR protein in normal pancreatic tissue is strongly expressed in the endocrine islet cells, but weakly expressed in exocrine acinar cells. The expression pattern of CAR in normal pancreatic tissue was further confirmed in another 10 normal pancreatic tissue specimens. This observation was different from the results reported by Korn and colleagues, in which CAR expression was particularly strong around pancreatic ducts. The ubiquitous expression of CAR suggested a broad tropism of rAdV; however, because of the variant CAR expression levels tissues might exhibit different levels of susceptibility to infection.
Another important issue in targeted delivery of therapeutic agents into pancreatic carcinoma is the route of rAdV administration. It affects both the efficacy of gene therapy and the systemic toxicity caused by transgene products released into the serum. In this study, we found that intraperitoneal injection of rAd-B7-mIL12 could lead to significantly higher peak concentrations of mIL-12 in serum than by intravenous or intramuscular injection. However, the decrease in serum concentration was more rapid after intraperitoneal injection so that after 3 days, the serum concentration of intraperitoneally injected animals was lower than that of intravenously or intramuscularly injected mice.
The peak concentration could be enhanced when rAd-B7-mIL12 was coinjected with RCAd, and the amount of increase was significant only after both adenoviruses were administered intraperitoneally. It was ineffective after intravenous injection, presumably because RCAd-augmented transgene expression required the expression of E1A, encoded by RCAd, in the same cell or adjacent cells (see Fig. 3B, panel e). This requirement was unlikely to be satisfied after intravenous injection because biodistributions of RCAd and rAd-B7-mIL12 might differ in animals. Further investigation is required to completely understand the underlying mechanisms of the observations shown in Fig. 5A.
As we have shown in the present study, both RCAd and VP16 could enhance RDAd-mediated gene delivery; however, the mechanisms of enhancement are different. RCAd helps RDAd replication in tumor cells by providing the E1A protein that RDAd lacks and that is required for replication. As a result, both DNA as well as protein levels of the transgene were found to be enhanced in treated cells and tissues. In contrast, VP16 appeared to have no effect on viral transduction or replication but could augment transgene expression levels in RDAd-infected cells, presumably by regulating its promoter. Further studies are required to determine the exact mechanism by which VP16 regulates transgene expression in RDAd-infected cells.
When used alone, both RCAd and VP16 at the doses used in this study had minimal toxicity in vitro in terms of cell killing, and in vivo as indicated by unchanged serum biochemical markers. This is backed up by the observation that VP16, combined with intraperitoneal injection of RCAd and/or RDAd, caused no obvious changes in tissue morphology of liver, kidney, spleen, and lung (data not shown).
In summary, the gene therapy approach developed in this study was based on two novel strategies. One was to combine therapeutic RDAd with an RCAd and VP16 at low doses to improve RDAd spreading in the tumor mass and RDAd-mediated gene transduction and expression efficiency. This strategy allowed adenoviral delivery of larger therapeutic genes, as compared with using replication-competent viruses, while still maintaining the safety properties inherent to replication-deficient adenoviruses. We think that this strategy could be generally used for rAdV-based therapies currently under development for other types of tumor.
The second approach we used in the present study was to target a therapeutic gene, PE40, to c-erb-B2, by fusing it with the respective monoclonal antibody and using it to treat pancreatic tumors, which commonly overexpress c-erb-B2, as we have determined by analyzing a number of normal and tumor pancreatic tissues and cell lines. As c-erb-B2 is found to be overexpressed in a variety of tumors a similar approach might be used in the treatment of abdominal metastases of other malignancies such as ovarian cancer. In addition, our approach can be modified to target other cell surface antigens commonly expressed in tumors, providing an attractive alternative in the development of novel cancer treatments.
Our new approach to gene therapy has proven to be effective for treating pancreatic tumors in mice. We suggest that our concept can be modified to work in the treatment of a variety of cancers.
Footnotes
Acknowledgments
The authors thank Chao Ge (Cancer Institute of Shanghai Jiaotong University) and Jing Sun, Jun Liu, Miaoying Yi, Yuanshan Lu, Jiawei Chen, and Zhaorui Yang (First People's Hospital, Shanghai Jiaotong University) for technical assistance and for providing materials for this research. This work was supported by a grant from the National Basic Research Project of China (2010CB529902), the National High-Tech R&D program (2007AA021202), the National Natural Science Foundation for Outstanding Youth (30325043 and 30428015), and the State Key Laboratory of Oncogenes and Related Genes (80-07-03).
Author Disclosure Statement
No competing financial interests exist for all authors.
*Address reprint requests to: Dr. Qian Huang.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.