
Abstract
The improvement of safety and titer of retroviral vectors produced in standard retroviral packaging cell lines is hampered because production relies on uncontrollable vector integration events. The influences of chromosomal surroundings make it difficult to dissect the performance of a specific vector from the chromosomal surroundings of the respective integration site. Taking advantage of a technology that relies on the use of packaging cell lines with predefined integration sites, we have systematically evaluated the performance of several retroviral vectors. In two previously established modular packaging cell lines (Flp293A and 293 FLEX) with single, defined chromosomal integration sites, retroviral vectors were integrated by means of Flp-mediated site-specific recombination. Vectors that are distinguished by different long terminal repeat promoters were introduced in either the sense or reverse orientation. The results show that the promoter, viral vector orientation, and integration site are the main determinants of the titer. Furthermore, we exploited the viral production systems to evaluate read-through activity. Read-through is thought to be caused by inefficient termination of vector transcription and is inherent to the nature of retroviral vectors. We assessed the frequency of transduction of sequences flanking the retroviral vectors from both integration sites. The approach presented here provides a platform for systematic design and evaluation of the efficiency and safety of retroviral vectors optimized for a given producer cell line.
Introduction
A number of distinct packaging cell lines have been developed on the basis of either murine or human cell lines. On stable integration of the retroviral vector into the host genome they are used for the production of infectious particles (Danos and Mulligan, 1988; Markowitz et al., 1988; Miller et al., 1991; Cosset et al., 1995; Miller and Chen, 1996; Ory et al., 1996; Ward et al., 2003). The titer of a retroviral vector produced from these packaging cell lines depends not only on the vector design but also on the copy number and the position effects mediated by its chromosomal integration. Genetic modification of these cells relies on unpredictable integration site distribution of the viral vector, making it impossible to investigate the properties of retroviral vectors independent of the variable influences of the integration site. Thus, rational design of vectors that takes advantage of beneficial effects of a given integration site on virus production is not possible. Accordingly, screening of appropriate integration sites that support optimal vector expression is still the state-of-the-art for establishment of producer cell lines—a highly time-consuming and laborious procedure.
The chromosomal site of vector integration is also of major importance because of the leakiness of termination of retroviral vector transcription (polyadenylation). Indeed, a suboptimal polyadenylation signal in the R region of the retroviral long terminal repeats (LTRs) results in the formation of read-through transcripts (Furger et al., 2001; Zaiss et al., 2002; Schambach et al., 2007). As much as 10% of viral transcripts are estimated to be a consequence of illegitimate read-through in retroviruses (Zaiss et al., 2002). This read-through activity is considered to be a mechanism that can lead to the transduction/activation of cellular oncogenes flanking the viral genome integration site (Swain and Coffin, 1992; Uren et al., 2005). For production of recombinant retroviral vectors the adventitious transduction of cellular genes by read-through activity is thus a safety concern. To exclude this, the specific vector integration site(s) in the chromosome of packaging cells would have to be characterized.
Two new modular retroviral packaging cell lines have been developed: Flp293A, with an amphotropic murine leukemia virus (MLV) host envelope (Schucht et al., 2006), and 293 FLEX, expressing the gibbon ape leukemia virus (GALV) envelope (Coroadinha et al., 2006). In each of these HEK293-based cell lines a single retroviral vector integration site was identified for its capacity to provide high retroviral vector production levels. Titers up to 2.5 × 107 infectious particles (IP)/106 cells in 24 hr were achieved from a single retroviral vector copy integrated into these highly active chromosomal sites.
A specific feature of these cells is the fact that the respective retroviral vector integration site is tagged with Flp recombinase target (FRT) sites and, moreover, linked to a sensitive selection system (Verhoeyen et al., 2001). This enables the efficient excision of integrated retroviral screening (tagging) vector and concomitant targeting by any (retroviral) vector of interest (Flp recombinase-mediated cassette exchange, RMCE). For both cell lines, cassette exchange is highly efficient and allows the rapid generation of recombinant virus with high and predictable titers. The schematic representation of the targeting reaction mediated by Flp in Flp293A and 293 FLEX cell lines is depicted in Fig. 1A–C. Apart from obvious advantages for highly controlled production of a given vector, these systems constitute an exceptional platform to systematically evaluate various vector compositions in defined chromosomal loci, allowing a direct comparison of vector performance and thus contributing to the development of a rational strategy for retroviral vector design. Further, because the 3′ elements flanking the retroviral vector integration site are known, a detailed investigation of vector-related read-through activities is feasible.
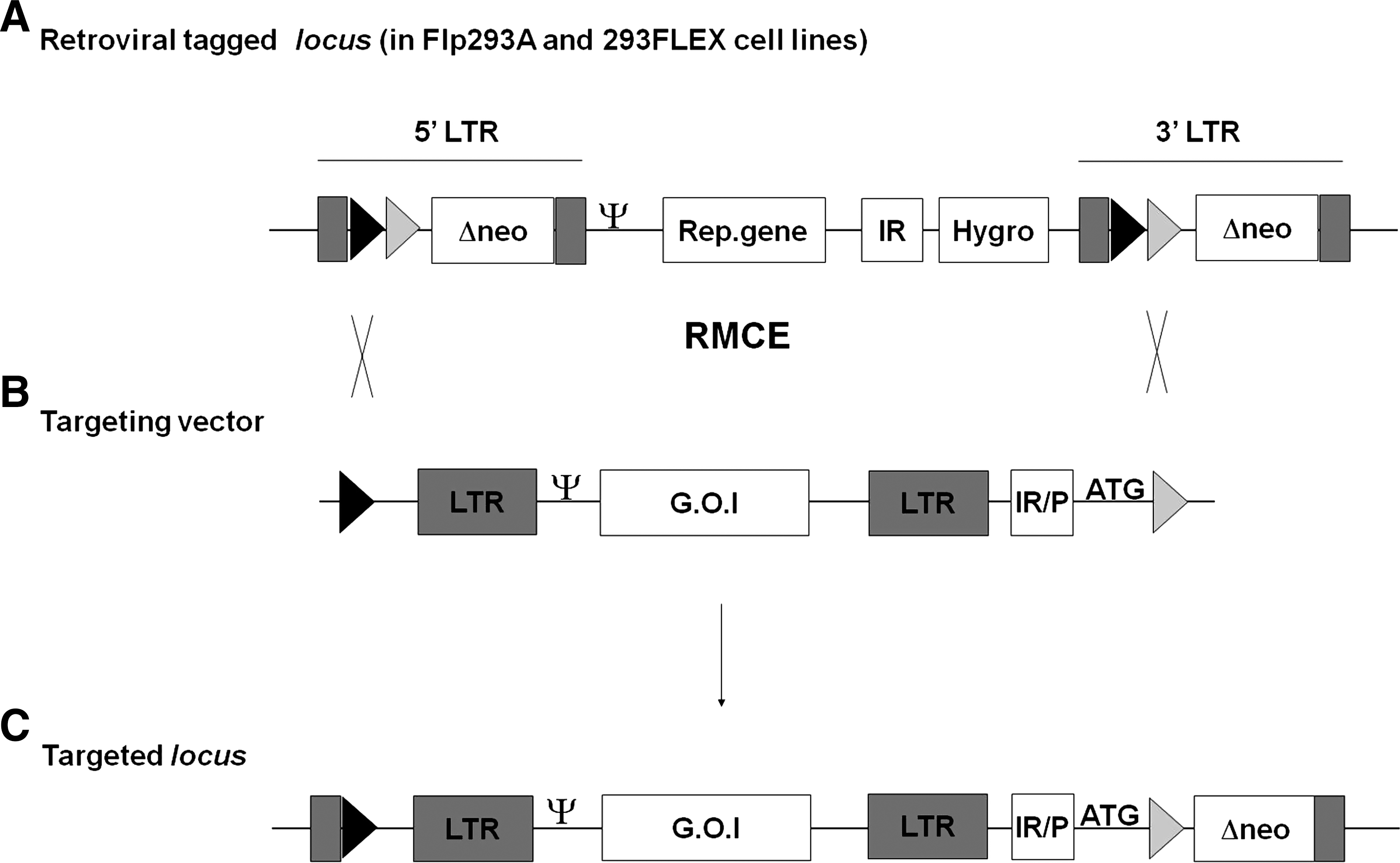
Vector targeting mediated by Flp recombinase-mediated cassette exchange (RMCE) in Flp293A and 293 FLEX. The tagged retroviral locus in Flp293A and 293 FLEX cells is represented (
The aim of this study was to evaluate retroviral vector design within the unique and defined chromosomal sites of modular cell lines on Flp-mediated integration. In particular, we tested the impact on retroviral production level of both 5′ promoter composition and vector orientation of the integrated retroviral genome. Further, we used the defined retroviral integration sites to assess the risk of the formation and transduction of specific read-through transcripts generated from retroviral vectors. Together, this knowledge will contribute to a rational design of retroviral vectors, thereby exploiting the properties of defined vector integration sites. We envision that the definition of optimal combinations of vector elements and integration sites will allow maximal production of vectors without compromising the biosafety needed for the implementation of these vectors in the clinic.
Materials and Methods
Plasmids
Targeting green fluorescent protein-expressing self-inactivating gammaretroviral vectors
Retroviral vectors were integrated into a precursor targeting vector (pEMTAR derived) containing FRT wild-type (F-WT) and FRT mutant (F-5) and an internal ribosomal entry site from encephalomyocarditis virus (EMCV IRES) (sense vectors) or a human phosphoglycerate kinase (PGK) promoter (reverse vectors). In all vectors, an ATG start codon is positioned upstream of the F-5 site to complement the ATG-deficient neomycin phosphotransferase gene on targeting. Sense and reverse targeting vectors differ in the orientation of the transcription of the retroviral cassette (convergent or divergent) with respect to transcription of the ATG-complemented neo gene on targeting event.
The sense targeting vectors: pRSV, pCMV, pSVe-RSV, and pMPSV are derived from pSRS11.SFGFPpre, pSCS11.SFGFPpre, pSERS11.SFGFPpre, and pSIN11.SFGFPpre, respectively, all previously described elsewhere (Schambach et al., 2006b). Briefly, the precursor vectors were cloned in the pEMTAR-derived vector in order to create targetable retroviral vectors. The vector names are modified from the original names in order to create more intuitive designations. The original names of the targeting vectors are pEMTARSRS11.SFGFPpre (pRSV), pEMTARSCS11.SFGFPpre (pCMV), pEMTARSERS11.SFGFPpre (pSVe-RSV), and pEMTARSIN11.SFGFPpre (pMPSV). pCMVe-MoMLV is derived from pE336 (Loew et al., 2010). The vector was modified into a targetable vector by cloning it into the pEMTAR-derived vector pTAR (Loew et al., 2010). pCMV-CPCol is derived from the targeting vector pBulletSintec 4 (Schucht et al., 2006) (referred to as pSINColVII). The elongation factor (EF)-1α internal promoter of pBulletSintec 4 was replaced with the human collagen promoter from the pCPColVII vector (kindly provided by O.W. Merten, Généthon, Evry, France).
The reverse targeting vectors: pPGK-MPSV and pPGK-SVe-RSV are derived from pSIN11.SFGFPpre and pSERS11.SFGFPpre, respectively, further modified by cloning the retroviral cassette into a pEMTAR-derived vector containing the PGK promoter as the regulatory element. The original designations of these vectors are pPGK-pEMTARSIN11.SFGFPpre (pPGK-MPSV) and pPGK-SERS11.SFGFPpre (pPGK-SVe-RSV). To generate pPGK-CMV-CPCol, the cassette containing the human collagen gene driven by the human collagen promoter was cloned into an EMTAR-derived vector containing the PGK promoter as the regulatory element driving expression of the neo gene in targeted cells.
Further details of all the vectors included in this study will be provided on request.
Mammalian cell culture
The modular packaging cell lines Flp293A (Schucht et al., 2006) and 293 FLEX (Coroadinha et al., 2006) were used for retroviral vector production on targeting via RMCE with the various targeting vectors described previously. These cell lines are based on HEK293 cells and provide a single chromosomal site tagged with two heterospecific and noncompatible FRT sites to allow Flp-mediated cassette exchange. Further, they carry constructs encoding the respective envelope and the gag-pol gene randomly integrated into the genome at an unknown copy number. NIH/3T3 cells (CRL-1658; American Type Culture Collection [ATCC], Manassas, VA) and TE671 cells (CRL-8805; ATCC) were used as target cells to titer the infectious retroviral vector particles produced from Flp293A and 293 FLEX, respectively. All cell lines were cultivated at 37°C in an humidified atmosphere with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, Munich, Germany) supplemented with 10% fetal bovine serum (BioWest, Nuaillé, France), 2 mM
Selection of targeted cells was performed with medium supplemented with G418 (1500 μg/ml) and ganciclovir (10 μg/ml). neo-transducing virus titration was performed with medium supplemented with G418 (1500 μg/ml).
Recombinase-mediated cassette exchange
For site-specific cassette exchange, Flp293A and 293 FLEX cells were seeded in a 6-well plate (3 × 104 cells/cm2) on day 0. On day 1, the cells were cotransfected with 2 μg of Flp recombinase-expressing vector (pFlpe; K. Maass [NYU School of Medicine, New York, NY], unpublished data) and 2 μg of targeting vector using GenePORTER 2 transfection reagent (Genlantis, San Diego, CA). Cells transfected only with the Flp coding vector and nontransfected cells were included as negative controls. Twenty-four hours posttransfection, the medium was replaced. On day 5, the cells were transferred to a 60-mm culture plate and cultivated in G418 and ganciclovir-containing medium to select for targeted clones. The selection was carried out for 14 days, during which it was ascertained that the cells in a negative control were dead. Putative RMCE targeted G418/ganciclovir-resistant clones were then picked and cultured.
Characterization of correctly targeted clones
G418- and ganciclovir-resistant single clones were evaluated for correct targeting by PCR. Genomic DNA was extracted accordingly to Ramirez-Solis and colleagues (1992) and used as template for PCR with the primer pair 5EMCV (5′-GGCAGCCAGTCGACGTTATTTTCCACCATATTGCCG-3′) and neorev2 (5′- GTCATAGCCGAATAGCCTCTCC-3′) in the case of sense targeting vectors and with the primer pair PGKFwd1 (5′-TCTCGCACATTCTTCACGTCC-3′) and neorev2 in the case of antisense targeting vectors.
Viral supernatant production and titration
Flp293A- and 293 FLEX-derived target clones were seeded at 4 × 104 cells/cm2 in 25-cm2 T-flasks with 3 ml of medium, ensuring a cell confluence of about 70% in 24 hr. The next day, supernatant was harvested, filtered (pore size, 45 μm), and used for infection of target cells on supplementation with Polybrene (8 μg/ml; Sigma-Aldrich). The cell number of the producer clones was determined.
Viral titration of green fluorescent protein (GFP) gene-carrying vectors generated on targeting of Flp293A and 293 FLEX was determined by flow cytometry. On day 0, NIH/3T3 and TE671 cells were seeded at 5 × 103 cells/cm2 in a 12-well plate. On day 1, NIH/3T3 and TE671 cells were infected with 300 μl of serial dilutions of viral supernatant generated from Flp293A- or 293 FLEX-derived clones, respectively. On day 2, the viral supernatant was replaced with fresh medium. On day 3, the infected cells were analyzed by flow cytometry. The titer was calculated on the basis of the percentage of GFP-positive cells. Infections that rendered 2–20% infected cells were considered for titer calculations.
Viral titration of collagen VII gene-carrying vectors on targeting of Flp293A and 293 FLEX was done by immune staining of infected cells. On staining, cells were analyzed by flow cytometry and the titer was determined. On day 0, NIH/3T3 and TE671 cells were seeded at 1 × 105 cells/cm2 in a 6-well plate. On day 1, NIH/3T3 cells and TE671 cells were infected with 1 ml of 1:2 and 1:10 dilutions of viral supernatant generated from Flp293A or 293 FLEX-derived clones, respectively. On day 2, the viral supernatant was replaced with fresh medium. On day 3, the cells were stained and the titer was determined. Briefly, the cells were washed twice with phosphate-buffered saline (PBS), fixed with 100 μl of 2% paraformaldehyde (PFA) solution, and again washed with PBS. The cells were stained with 100 μl of 1:300 diluted primary antibody solution (anti-human collagen type VII, C6805; Sigma-Aldrich) in 0.5% Triton–PBS and incubated for 30 min on ice. After incubation the cells were washed three times with 0.5% Triton–PBS. The secondary goat anti-mouse IgG antibody (labeled with fluorescein isothiocyanate [FITC]; Dianova, Hamburg, Germany) was applied (diluted 1:400) and the cells were incubated for 20 min on ice. After incubation, the cells were washed twice with 0.5% Triton–PBS and finally resuspended in PBS–2% FBS FACS buffer.
Flow cytometry
Flow cytometric analysis was performed on a FACSCalibur (BD Biosciences, San Jose, CA). For titration of GFP-expressing virus, the cells were washed, trypsinized, and stained with propidium iodide (50 μg/ml) to exclude dead cells.
neo-transducing virus titration
Viral supernatants were harvested from targeted 293 FLEX and Flp293A cells and used to infect TE671 or NIH/3T3 cells, respectively. Briefly, TE671 and NIH/3T3 cells were seeded at 1.0 × 104 cells/cm2 in a 6-well plate (day 0). On day 1, the cells were infected with 1 ml of serial 10-fold dilutions of viral supernatant. On day 2, the cells were transferred to a 60-mm culture dish in the presence of medium supplemented with G418. Fourteen days after, the number of G418-resistant cells was determined and the neo titer was calculated.
Results
Reuse of a defined chromosomal locus of a modular packaging cell line for evaluation of retroviral vector performance
Efficiency of viral RNA transcription is dependent on the repertoire of transcription factors provided by the packaging cell line (so-called trans factors) and on cis effects. The latter comprise regulatory elements within the vector sequence, genetic elements, as well as epigenetic organization status of the chromosomal integration site of the vector. To compare the performance of various retroviral vectors and their dependence on the chromosomal surroundings of the integration site, a panel of various gammaretroviral vectors (Fig. 2) was included in this study. These vectors were targeted into the tagged single-copy chromosomal loci of the modular packaging cell lines 293 FLEX and Flp293A (Coroadinha et al., 2006; Schucht et al., 2006).
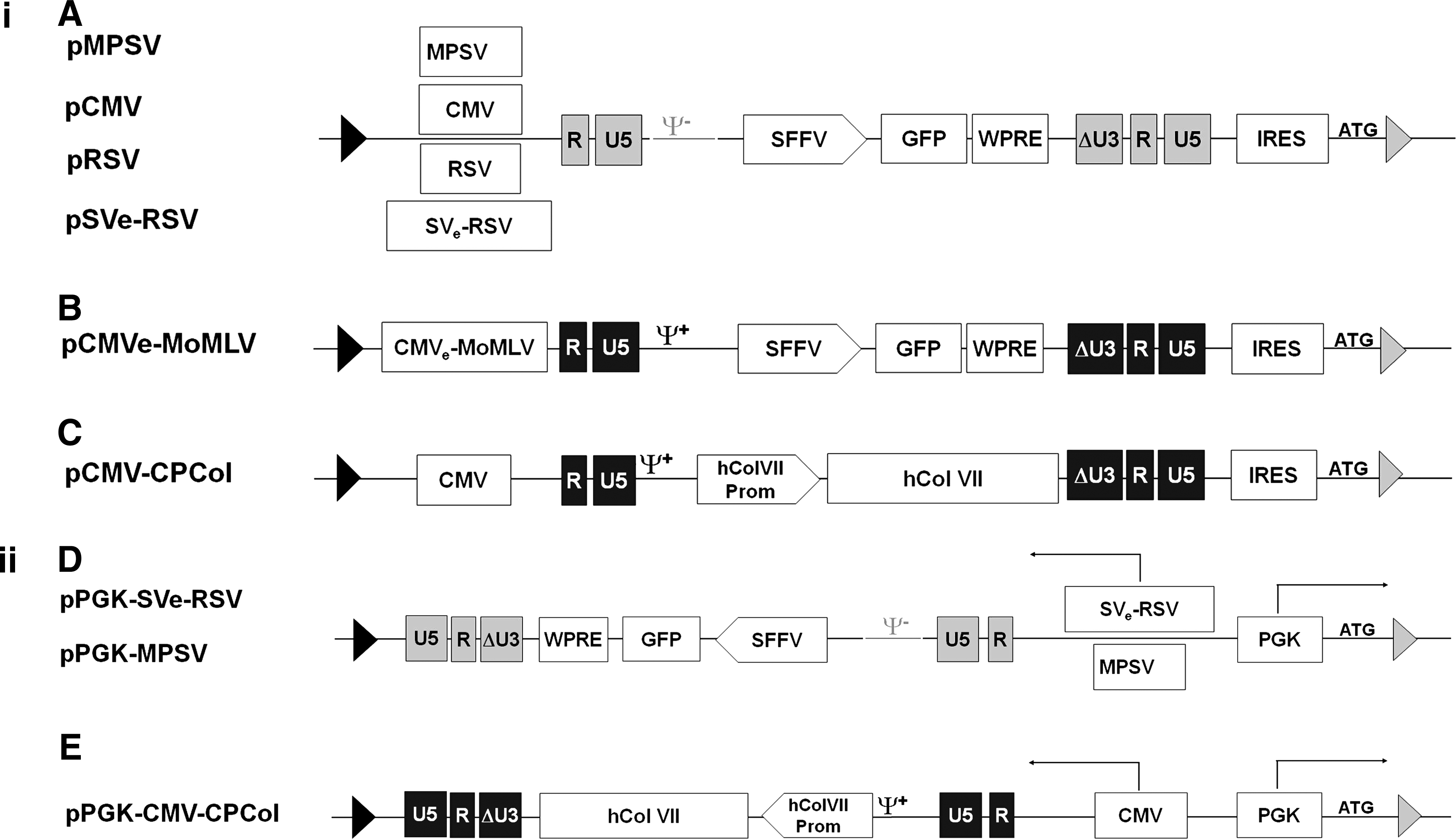
Schematic representation of targeting self-inactivating (SIN) gammaretroviral vectors used in this study. The retroviral vectors are flanked by two noninteracting Flp recombinase target (FRT) sites, F-WT and F-5, depicted as black and gray triangles, respectively. Selection of the correctly targeted clones is ensured by the presence of the internal ribosomal entry site from encephalomyocarditis virus (EMCV IRES), in the case of sense vectors (
The retroviral vector integration site of these two cell lines was mapped, using a nonrestrictive linear amplification-mediated (LAM)-PCR protocol (Gabriel et al., 2009). For Flp293A the tagged locus is located on the short arm of chromosome 12, either in the first intron of the DDX11 gene (p11.21, position 31227164) or the DDX12 gene (p13.31, position 9600360), reversely oriented in relation to endogenous gene transcription. Because these genes are highly homologous, both positions are possible. For the 293 FLEX cell line, the tagged locus is positioned in an intergenic region 100 kb upstream of the gene ARRDC3 on chromosome 5 (q14.3, position 90781745). After Flp-mediated targeting of the retroviral vectors in Flp293A and 293 FLEX cells, G418-resistant clones were isolated and targeting was confirmed by PCR (data not shown). Virus production from the targeted cells was determined. In agreement with previous findings (Coroadinha et al., 2006; Schucht et al., 2006), the virus production levels of clones generated on cassette exchange with a given vector were highly homogeneous. This is exemplified in Fig. 3 for the vectors pSVe-RSV and pCMV after targeting.
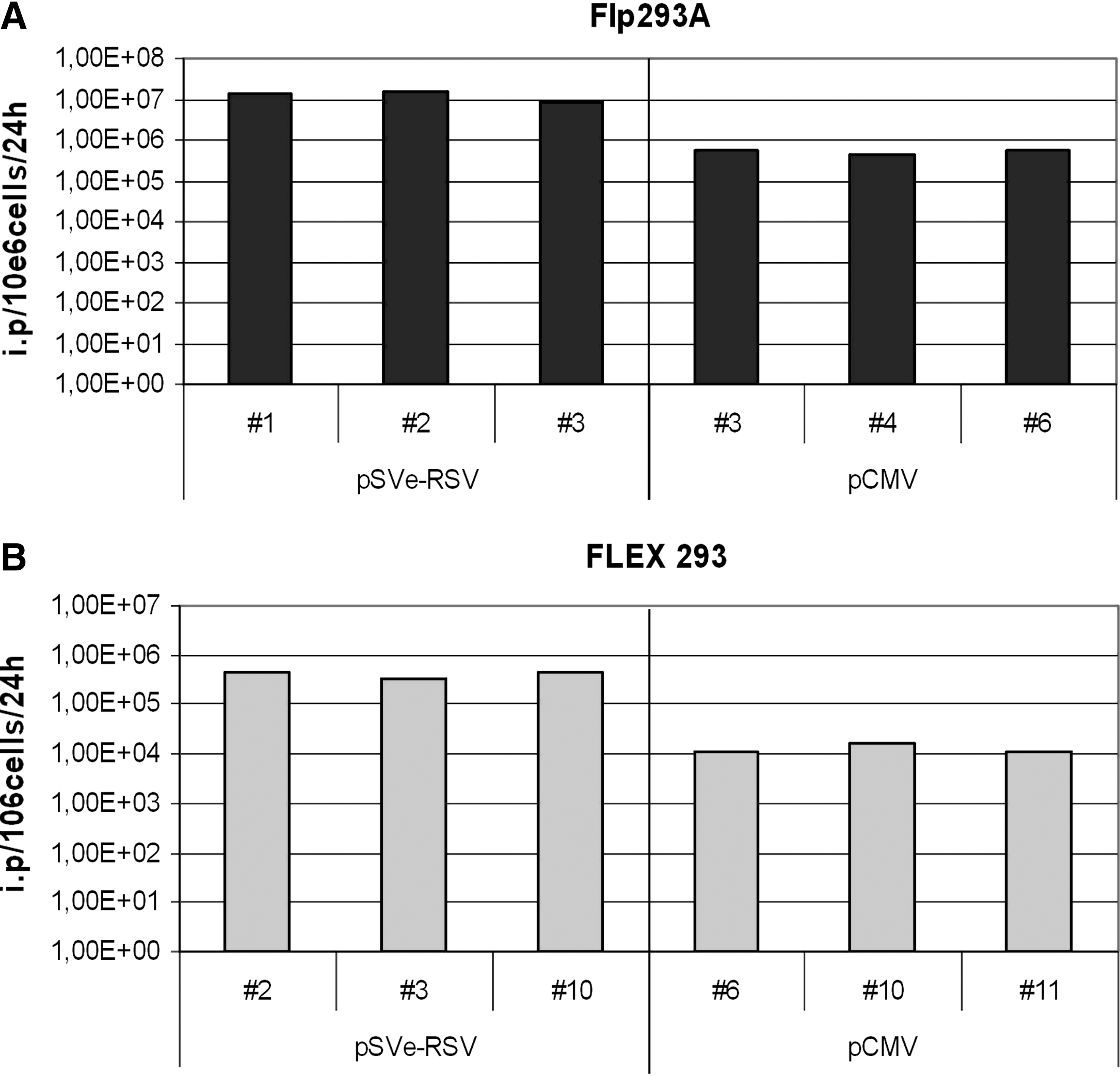
Analysis of virus production from independent cell clones obtained on targeting of Flp293A (
Impact of regulatory elements
Among other elements, the nature of the promoter that drives retroviral vector transcription is important for the titer. To compare various commonly used promoter elements in the defined chromosomal context of the modular cell lines Flp293A and 293 FLEX, we used self-inactivating (SIN) gammaretroviral vectors as displayed in Fig. 2i, A and B (for the sake of simplicity, the name of each vector is deduced from the promoter within the 5′ LTR). Four of these SIN vectors differ only in 5′ promoter/enhancer elements used for driving the viral genomic mRNA (Schambach et al., 2006b). Both native promoter sequences (myeloproliferative sarcoma virus [MPSV], cytomegalovirus [CMV], and Rous sarcoma virus [RSV]) and a combination of simian virus 40 enhancer and RSV promoter element (SVe-RSV) were employed (Fig. 2i, A). Further, we included a Moloney murine leukemia virus (MoMLV)-based SIN vector in which the endogenous U3 sequence in the 5′ LTR was replaced by a CMV enhancer–MoMLV composite promoter (CMVe-MoMLV) (Fig. 2i, B). Common to the vectors included in this study is the internal cassette composed of the spleen focus-forming virus (SFFV) U3 sequence that drives the GFP reporter gene.
High clonal homogeneity was observed for all cell clones generated on targeting of the same retroviral vector (Fig. 3). However, we found up to 15-fold differences in retroviral titers when the five different vectors were compared in the same locus. Figure 4 depicts a summary of the titers obtained from clones generated on targeting of Flp293A and 293 FLEX with the five vectors.
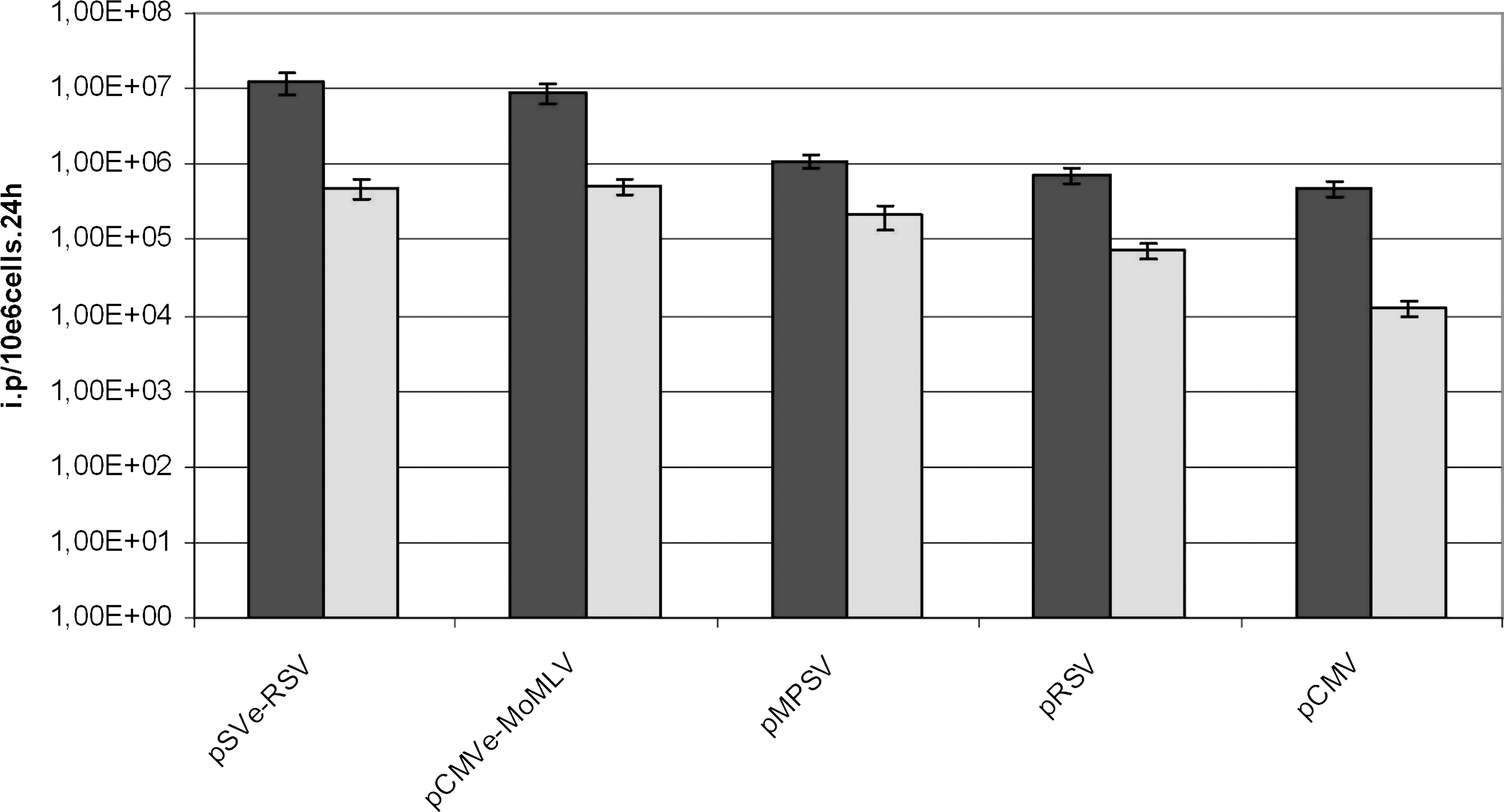
Influence of the 5′ LTR promoter on virus titer from targeted retroviral modular cells. The gammaretroviral SIN vectors as depicted in Fig. 2i, A and B were targeted in the Flp293A cell line (dark gray bars) and 293 FLEX cell line (light gray bars). The supernatant from three different cell clones from each targeting in the Flp293A and 293 FLEX lines was used to infect NIH/3T3 and TE671 cells, respectively. Virus titrations were done by assessing the number of GFP-expressing cells by FACS. Average titers were calculated on the basis of at least three measurements of GFP expression on infection, using independent cell clones obtained after RMCE.
Interestingly, the hierarchy of performance of the 5′ LTR-modified retroviral vectors was the same in both packaging cell lines. The composite promoter vectors pSVe-RSV and pCMVe-MoMLV provided the highest titers: about 1 × 107 IP/1 × 106 cells in 24 hr in Flp293A and about 7 × 105 IP/1 × 106 cells in 24 hr in 293 FLEX. pRSV and pCMV gave rise to significantly lower titers in both cell lines.
Impact of the orientation of vector integration
We further asked whether the integration sites in Flp293A and 293 FLEX cells support efficient vector production for both orientations of the retroviral vector according to Fig. 5A. For this study, the two GFP-carrying SIN gammaretroviral vectors pMPSV and pSVe-RSV (Fig. 2i, A) were chosen and integrated into a targeting vector in reverse orientation. To activate the neo gene on integration, the human PGK promoter was used. This provided the targeting plasmids pPGK-MPSV and pPGK-SVe-RSV (Fig. 2ii, D). In this study we further included an MoMLV-derived collagen VII gene-transducing vector, pCMV-CPCol (Fig. 2i, C). In its reverse counterpart, pPGK-CMV-CPCol (Fig. 2ii, E), genomic viral RNA transcription is driven by a CMV promoter. Interestingly, the vector orientation had different effects in the two integration sites. In the 293 FLEX site, both orientations resulted in either similar titers or an increase in titer from the reversely oriented collagen VII-containing vector when compared with its sense counterpart (Fig. 5B). In contrast, in the Flp293A site, a significant lower titer was observed in all reverse targeting vectors (Fig. 5C). For the vector pPGK-SVe-RSV a difference of more than 7-fold was observed when compared with pSVe-RSV, whereas for pPGK-MPSV a reduction of about 13-fold was observed when compared with the sense counterpart pMPSV. An even more severe reduction in titer was observed when comparing the sense and reverse collagen-expressing vectors in Flp293A. This shows that although the integration site of 293 FLEX cells supports both orientations equally, in Flp293A cells the sense orientation is highly favored.
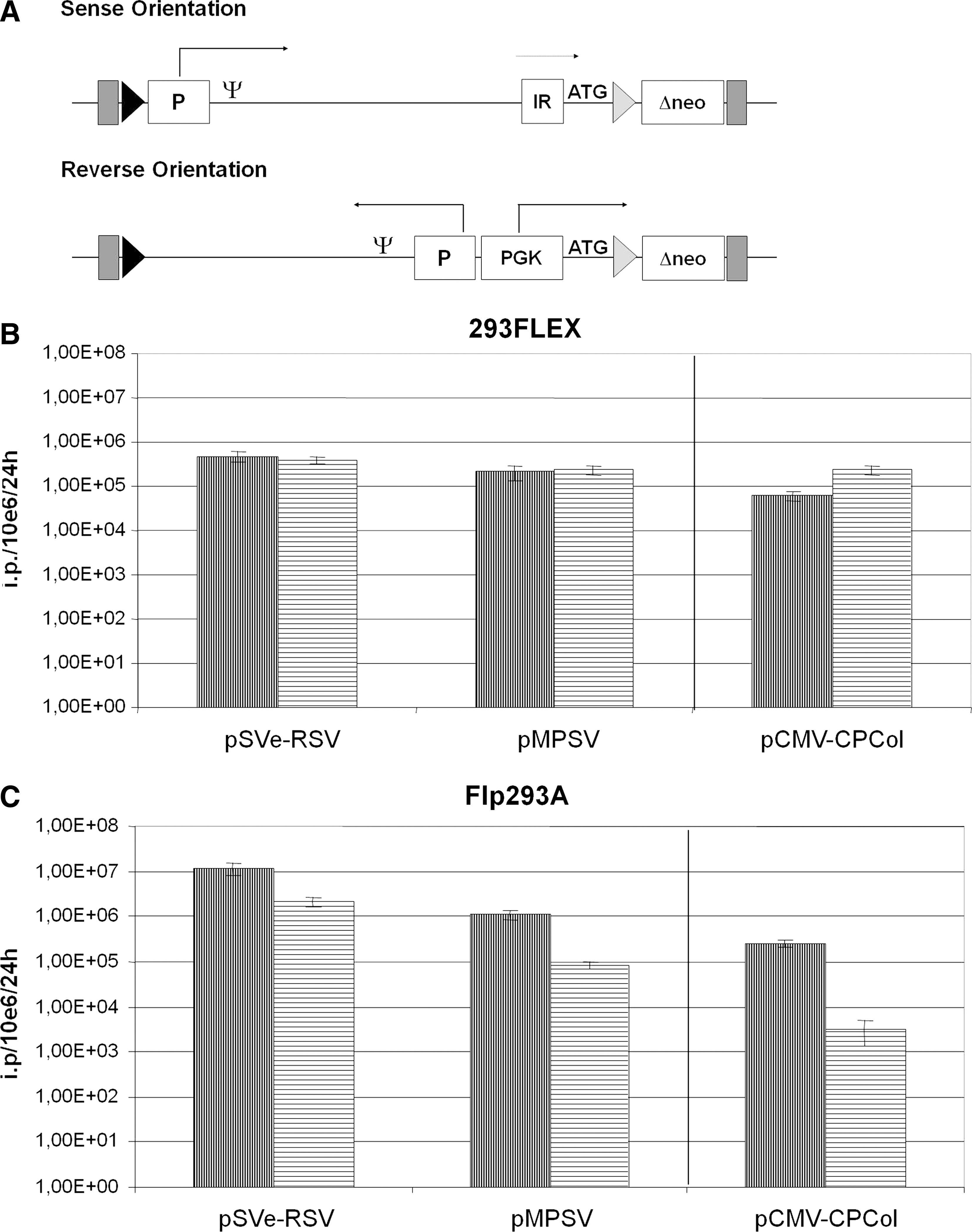
The impact of orientation of retroviral transcription on infectious viral production from Flp293A and 293 FLEX cell lines. (
Evaluation of packaging of read-through transcripts in modular packaging cell lines
In the modular producer cell lines Flp293A and 293 FLEX, the downstream region of the 3′ LTR of the targeted vectors is defined. It is composed of the FRT F-5 site followed by the neomycin phosphotransferase gene (neo) and the residual 3′ LTR sequences. These elements were used to identify correctly targeted retroviral vectors. On targeting of a retroviral vector in sense orientation into the tagged sites, the vector is flanked at its 3′ end by the EMCV IRES element followed by the neo and an inverted repeat-deleted viral LTR, as depicted in Fig. 1C. Because the termination of retroviral vector transcripts is provided by a weak polyadenylation signal present in the R region of the viral LTR, we asked whether this configuration would result in transduction of the downstream sequences. As these events are expected to be comparably rare, we exploited the unique situation in the modular packaging cell lines and determined the ability of the previously described producer clones to transduce the neo gene (“read-through titer”). Packaging of these read-through transcripts on infection and integration should render neomycin resistance. In particular, we intended to investigate whether the read-through events are strictly dependent and directly correlated with the strength of a certain promoter or with a certain promoter composition.
To evaluate the read-through frequency, viral supernatants from targeted 293 FLEX and Flp293A cells were used to infect TE671 and NIH/3T3 cells, respectively. These read-through titers associated with the tested vectors in 293 FLEX and Flp293A are summarized in Tables 1 and 2. The number of G418-resistant cells reflects the frequency of read-through and successful transduction of the neo gene during virus production. The read-through titers were compared with the number of GFP-expressing infected cells, which were taken as a measure of total infectious viral particle production (Tables 1 and 2). The ratio between the read-through titer and the total infectious particle titer gives a quantitative measure of read-through occurrence frequency.
neo
neo titer: G418-resistant particles/1 × 106 producer cells in 24 hr. Between three and eight independent measurements for each targeting group were considered. For the sense vectors, neo titer reflects the read-through activity.
Infectious particles titer: GFP-transducing particles/1 × 106 producer cells in 24 hr.
neo titer/infectious particles: This ratio represents the frequency of neo transduction events per total number of infectious particles produced.
neo
Abbreviation: ND, not determined.
neo titer: G418-resistant particles/1 × 106 producer cells in 24 hr. Between three and eight independent measurements for each targeting group were considered. For the sense vectors, neo titer reflects the read-through activity.
Infectious particles titer: GFP-transducing particles/1 × 106 producer cells in 24 hr.
neo titer/infectious particles: This ratio represents the frequency of neo transduction events per total number of infectious particles produced.
The read-through titers produced by the clones targeted with the various sense vectors are considerably heterogeneous, with a two orders of magnitude difference between the two cell lines tested. Namely, the 293 FLEX cell line targeted with pCMVe-MoMLV and pCMV produced about 12 to 16 neo-transducing IP/106 cells in 24 hr, whereas the 293 FLEX cell line targeted with pSVe-RSV, pMPSV, and pRSV produced 370 to 922 neo-transducing IP/106 cells in 24 hr. Similarly, Flp293A-derived cells targeted with pCMVe-MoMLV and pCMV were associated with a read-through frequency 30- to 100-fold lower than the read-through frequency observed for the cell line targeted by pSVe-RSV, pMPSV, and pRSV. It is noteworthy that efficiency of read-through transduction was not correlated with the GFP-transducing infectious viral particles produced by the constituent vectors in both of the cell lines tested: among the vectors with low neo titers, pCMVe-MoMLV was one of the two vectors with the highest infectious particle production. In contrast, pCMV also showing low neo titers was the vector that showed the lowest infectious particle production capacity. At the same time, for the vectors that gave rise to the highest infectious particle titers, pSVe-RSV and pCMVe-MoMLV, the efficiency of read-through transcript transduction was found to be low. In 293 FLEX one neo-transducing particle was generated for every 810 or 15,000 infectious particles produced from cells targeted with pSVe-RSV and pCMVe-MoMLV, respectively. Interestingly, the frequency of read-through transcripts generation was maximally 2- to 4-fold increased in Flp293A cells, although this cell line provided an up to 40- to 50-fold increased GFP transduction capacity.
In general, the maximal size of naturally occurring retroviruses is about 9.5 kb. Viral RNAs that exceed this length are found to be compromised for packaging and transduction. Hence, theoretically, long read-through transcripts should be less efficiently packaged than the shorter, correctly polyadenylated transcripts. We evaluated the efficiency of neo transduction from cells targeted with the vector pCMV-CPCol (Fig. 2i, C). This cDNA comprises about 9 kb; on correct polyadenylation at the 3′ R region a transcript of 10.5 kb is expected, whereas illegitimate read-through transcripts would result in a 12.5-kb RNA. As expected, 293 FLEX as well Flp293A cells targeted with pCMV-CPCol are related to low levels of read-through transcript generation, producing, on average, 12.5 and 40.8 neo-transducing particles per 106 cells in 24 hr, respectively, and therefore ranging at the lower level. These results also show that the increase in RNA length per se cannot further reduce the neo titer, indicating that additional mechanisms are involved.
Overcoming the formation of neo-transducing read-through transcripts by targeting the retroviral cassette in reverse orientation
Accidental transduction of the neo gene does not represent a critical issue per se and indeed many therapeutic vectors use the neo gene as a cotransduced selection marker. However, apart from potential antigenicity, the neo transduction from modular cell lines is accompanied by an unwanted transfer of the 3′ LTR elements (remaining sequences from the tagging construct after a targeting event). This illegitimate transduction thus represents the transfer of sequences with promoter/enhancer properties with the ability to restore LTR activity on infection. Indeed, it has been shown that these promoter/enhancer elements have the potential to deregulate genes in the vicinity of the vector integration site (Hacein-Bey-Abina et al., 2003).
Although the frequency of putative insertional mutagenesis events triggered on infection with the read-through-derived particles is rather low, we intended to improve the safety of these cellular systems for retroviral vector production. Thus, we evaluated whether a reverse orientation of retroviral vectors in the previously tagged integration sites would efficiently overcome transduction of the residual sequences (i.e., neo and the LTR) provided by the modular cell lines. For this purpose, the vectors pSVe-RSV, pMPSV, and pCMV-CPCol were chosen. Read-through titers from targeted cells with reverse-oriented vectors were determined and compared with the read-through titers produced by the sense counterparts (Tables 1 and 2).
As expected, a significant reduction of neo-transducing particles was observed for the reverse-oriented vectors. In the case of the reverse GFP-carrying vectors targeted into the 293 FLEX cell line, the ratio of neo-transducing particles to total infectious particles was only 1 in 1 × 105 and less than 1 in 4 × 105 for pPGK-MPSV and pPGK-SVe-RSV, respectively. In the case of reverse-oriented vector pPGK-CMV-CPCol no G418-resistant colonies were detected, indicating that less than 1 in 2 × 105 of infectious particles transduced the neo-containing transcript. A significant 15-fold reduction was observed in the case of Flp293A targeted with the collagen-encoding reverse vector. Thus, we show that in modular packaging cell lines 293 FLEX and Flp293A, expression of the reverse-oriented retroviral vectors is efficiently supported and significantly reduces transduction of the neo gene and 3′ LTR-containing transcripts.
Discussion
The modular packaging cell lines Flp293A and 293 FLEX represent universal tools for the production of any gammaretroviral vector by application of the recombinase-mediated cassette exchange technique. Because of the controlled genetic modification mediated by targeting of a characterized integration site, a predictable, fast, and safe production of viral vectors is possible, thereby overcoming limitations of strategies that rely on random integration of the viral genome. Both modular packaging cell lines are derived from the same cell line (HEK293), but are distinguished by the integration sites of the retroviral construct. The absolute titer cannot be compared between the cell lines because Flp293A produces the amphotropic envelope and 293 FLEX packages the virus with the GALV envelope. Nevertheless, the relative amount of viral production can be compared. This allowed the performance evaluation of various retroviral vectors in each of the two integration sites. Among the variables affecting the viral titer in a given cell, the cell's repertoire of transcription factors (trans effects) defines the overall activity of specific promoters. These trans effects should act independently of chromosomal integration. In addition to these “global” effects, any integration site in the genome may be affected by positive and/or negative effects mediated by genetic and epigenetic elements. These cis effects are site specific and may significantly modulate the transcription efficiency of cassettes that are integrated into a specific site.
The consistent graduation obtained after targeting various vectors differing in their 5′ promoters into the chromosomal sites of Flp293A and 293 FLEX (Fig. 1A) suggests that a corresponding set of cellular factors determines the titer in both cell lines. This is expected for typical trans effects such as the availability of certain transcription factors required for promoting transcription of the individual promoter/enhancer combinations. Also, cis factors acting on both integration sites in Flp293A and 293 FLEX cells would give rise to a consistent performance of vectors. In this respect, the tagging procedure applied for identification of the integration sites and in particular the common flanking 3′ LTR sequences of the tagging virus might contribute to this consistency. Interestingly, a totally different hierarchy was found if the vectors were evaluated in a mouse-derived modular packaging cell line that was established by the same screening procedure (Loew et al., 2010). This suggests that the trans effects have a strong influence in determining viral vector production levels from stable helper cell lines.
Besides this consistent behavior of vector performance at the two chromosomal loci, an integration site-specific modulation is thought to result in a locus-specific “fine-tuning” of the transcription regulatory elements. An example is the vector pCMV in comparison with the pRSV vector: Although these vectors show comparable performances in the Flp293A cell line the CMV vector is 5.5-fold less active in the 293 FLEX cell line. Conversely, pSVe-RSV and pMPSV vectors behave similarly in 293 FLEX (5 × 105 and 2 × 105 IP/1 × 106 cells in 24 hr, respectively) but display a more than 10-fold difference in titer in Flp293A. We conclude that these different performances are a consequence of locus-specific cis effects on the respective promoter elements. In this respect, the CMV promoter is either negatively influenced by the chromosomal surroundings of the 293 FLEX locus, or positively affected by the surroundings in the Flp293A cell line. This gives evidence that, indeed, optimal combinations of specific integration sites and promoter content of a retroviral vector must be defined in order to maximize the level of recombinant virus produced by those systems.
Notably, the significant difference in the performance of the vectors with the highest and the lowest viral titers (pSVe-RSV and pCMV, respectively) of 25- to 33-fold in Flp293A and 293 FLEX was not obtained in transient production experiments (Schambach et al., 2006b). Nevertheless, in both production systems (transient and stable production), vector pSVe-RSV performed best and vector pCMV performed worst. This supports the view that trans-acting factors in HEK293-derived cells (e.g., transcription factors) govern this hierarchy. In addition, this gives evidence that in transient production systems, the simultaneous expression of multiple copies of the retroviral genome can mask the real impact of the different heterologous 5′ LTR promoter on the full-length transcript production and concomitantly on the infectious viral titers.
Further, our study clearly shows that transcription of a targeted vector in either one or the other orientation is integration site dependent. Whereas in Flp293A the sense orientation is highly favored (relative to the reversed vector), in 293 FLEX cells efficient production is supported on reversion of the vector to the same extent as for the sense vector.
We assume that cross-talk between cellular and vector elements (cis effects) plays a major role in determining the expression level of a certain integrated cassette. The tagging vector integration site of Flp293A has been mapped to chromosome 12 in the first intron of the DDX11 or DDX12 gene, with reverse orientation to the DDX transcript. Despite potential antisense transcripts in Flp293A, high expression of targeted retroviral vector requires its orientation in antisense to the endogenous transcript. A possible explanation concerns the promoters of the endogenous gene (DDX11/DDX12) and the vector. In antisense orientation, the endogenous DDX11/DDX12 gene and the retroviral vector are divergent and the promoters are separated by 10 kb of intervening sequence. A different scenario is met for the 293 FLEX cell line. As referred to previously, the tagged site is positioned in an intergenic region of chromosome 5, 100 kb upstream from the ARRDC3 gene. In this integration site, we determined no preference for the sense versus reverse orientation. We hypothesize that this might be due to the absence of interfering promoters or endogenous transcriptional activity in the vicinity of the tagged site.
Together, our study shows that retroviral titer can be maximized by acknowledging the specific requirements of the nature of the retroviral vector integration site in the producer cell line, suggesting that the chromosomal locus can modulate the performance of specific regulatory elements presented in the integrated viral vector. Thus, to achieve an optimal titer, the demands of the respective integration site must be considered. Accordingly, only certain vectors/promoters can provide satisfactorily high titers in a given integration site.
Retroviruses have been shown to acquire and transduce cellular genes. This can be accomplished by accidental packaging of cellular RNAs devoid of any retroviral sequences. This process is designated retrofection (Linial, 1987; Levine et al., 1990; Lum and Linial, 1998) and has been shown to occur at frequencies of 10−6. The frequency of retrofection must be considered as the basal transduction level inherent to any retroviral transfer. More relevant for accidental transduction are cellular genes that are located next to retroviral integration sites. This is the initial event that can lead to the capture of proto-oncogenes (Swanstrom et al., 1983; Herman and Coffin, 1987; Swain and Coffin, 1992; Schwartz et al., 1995; Muriaux and Rein, 2003). Illegitimate read-through of viral RNA is supposed to be one mechanism followed by copackaging and subsequent recombination with a correctly processed RNA. To exclude transduction of proto-oncogenes by this mechanism, the proximal 3′ region of a retroviral vector integration site in the packaging cell line must be confirmed to be devoid of critical sequences. For the retroviral integration sites of Flp293A cells and 293 FLEX cells the proximal 3′ region is constituted by elements used for the establishment of the integration site, including an additional polyadenylation signal provided by the residual 3′ LTR. In this respect, on transcription of a sense vector transduction of cellular neighboring sequences can be largely excluded for both of the cell lines. Further, because of the chromosomal localization of 293 FLEX tagged sites, putative read-through in either direction would not lead to expression of a functional protein in the infected cells.
Despite the high level of safety of modular packaging cells with respect to the adventitious transduction of cellular sequences, an inherent property of these cellular systems is the potential read-through transcription and packaging of sequences downstream from the targeted retroviral vector (composed of the neomycin phosphotransferase gene and retroviral LTR) that constitute the remaining sequences of the tagging vector on a targeting event. This specific feature could be used to determine the extent of read-through capacity of various retroviral vectors targeted to these specific loci simply by the quantification of G418-resistant cells that arise on infection. On comparing the neo transduction frequency of the vectors used, we observed that the frequency of neo transduction did not correlate with the GFP titer. The reason for this is not obvious. One explanation for this might be that the transcription termination is affected by specific DNA configurations promoted by the regulatory sequences presented in a certain targeted vector. Such regulatory sequences might positively or negatively affect the recognition of transcription termination signals presented at the 3′ end of the retroviral vector. Alternatively, unique genetic or structural elements inherent to both integration sites might provoke this inconsistency.
Because suppression of transcriptional read-through into downstream sequences can be regarded as an improvement, vectors such as pCMVe-MoMLV targeted into the modular cell lines constitute a better choice in terms of biosafety (Tables 1 and 2). Moreover, incorporation of sequences that improve transcript termination, such as upstream sequence elements (Schambach et al., 2007), can be regarded as a benefit for the safer production of therapeutics by such vector systems. It has been shown that promoters derived from human genes, such as EF-1α and PGK, show a lower potential to activate neighboring promoters than virally derived promoters (Zychlinski et al., 2008).
The safety of the production system presented in this work has also been shown to increase on targeting of retroviral vectors in reverse orientation. This strategy is of great value for the minimization of the production of viral transcripts that are formed on read-through events of the neo gene and LTR sequences in the targeted cells.
In the reverse orientation, read-through of the viral transcript cannot contribute to neo gene transduction. The low frequency of neo transduction is in the same range as has been reported for unspecific retrofection and in this respect must be considered the natural basal level of transduction of cellular sequences, independent of the orientation of the retroviral vector.
Although the integration of reverse-oriented vectors represents a beneficial feature for increasing the safety of retroviral vectors (concerning the specific characteristics inherent to these modular cell systems), it is of note that the performance of these reverse retroviral vectors is strictly dependent on the chromosomal locus. A similar conclusion is also supported by results obtained in another cellular background (Loew et al., 2010).
To achieve the production of vector particles meeting the required safety levels for the translation of these cellular systems into the clinic, the incorporation of sequences that improve transcription termination of the retroviral tagging sequence is an alternative strategy (Schambach et al., 2007). We anticipate that the read-through titer of sense vectors targeted in modular cell lines can be reduced by implementing additional polyadenylation sequences downstream of the viral genome, without a significant decrease in viral titer.
The data show that a systematic comparison of vector constructions is feasible in modular producer cell lines. Moreover, the data also suggest that the next generation of helper cell lines should have additional features: in particular, they should provide maximal flexibility with respect to supporting vector sequences and also chromosomal surroundings that do not lead to unpredictable effects such as oncogenic transfer or antigenic effects. As RMCE implies that the site of interest is tagged a priori by specific sequence motifs, we envision that technologies for site-directed modifications of mammalian genome, for example, zinc finger recombinase-based technologies, could be used to direct FRT docking sites to specific loci of interest. The tagging of a well-characterized defined locus and its exploitation by means of RMCE provides the next step toward the establishment of safer cell lines suitable for clinical-grade production of therapeutic vectors, representing a particular case of production of proteins with biological/therapeutical significance.
Footnotes
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Wi 2624, REBIRTH), the Bundesministerium für Bildung und Forschung (FKZ0313940), and the EU (Clinigene, LSHB-CT-2006-018933; Flpflex, MEST-CT-2004-504990). L.G.N. acknowledges the financial support received from Fundação para a Ciência e Tecnologia, Portugal (SFRH/BD/22081/2005). In addition, A.S. and C.B. were supported by EU (PERSIST) and by the Else Kröner Foundation.
Author Disclosure Statement
No competing financial interests exist.